
各論
Ⅰ.遺伝性大腸癌の概要(Outlines of Hereditary Colorectal Cancer)
1 基本的事項
- 遺伝性大腸癌の占める割合は,全大腸癌の約5%である。大腸癌患者の約30%は遺伝的素因があると考えられている。
- 原因遺伝子が同定されている代表的な遺伝性大腸癌として,家族性大腸腺腫症(familial adenomatous polyposis:FAP)やリンチ症候群がある。原因遺伝子によって,大腸癌発症リスクが異なり,合併する腫瘍の種類や頻度もさまざまである。
- FAPやリンチ症候群などの常染色体優性遺伝形式をとる大腸癌の発症では,原因遺伝子の片側のアレルに先天的に病的バリアント(サイドメモ1:バリアント,生殖細胞系列バリアントと体細胞バリアント)を有している状態に,対側のアレルにも機能喪失を引き起こす変化がtwo-hitとして大腸の上皮細胞に後天的に加わることでがん化すると考えられている。
- 生殖細胞系列において原因遺伝子の病的バリアントが検出されている大腸癌であれば,家族集積性に関係なく遺伝性大腸癌と定義される。遺伝性大腸癌の代表的な疾患は,FAPやリンチ症候群である。
- 家族性に集積を認める大腸癌の中には,原因遺伝子の病的バリアントが見つからないこともある(各論Ⅲ 2-2):家族性大腸癌タイプX参照)。
- 全大腸癌の約30%3,4)は,遺伝的素因のある大腸癌と考えられている(図1)。遺伝性大腸癌の全大腸癌に占める割合は,約5%5)である。
- リンチ症候群の全大腸癌に占める頻度は,これまで欧米の報告では2~4%6,7)と推定されている。
- 近年の全大腸癌を対象としたマイクロサテライト不安定性(microsatellite instability:MSI)検査,もしくはミスマッチ修復タンパクの免疫染色によるユニバーサルスクリーニング(各論Ⅲ 2-1):診断の流れ参照)から遺伝学的検査(サイドメモ1:遺伝学的検査)を行った結果,欧米から2.4~3.7%8,9),本邦からも1%未満10,11)と報告されている。
- FAPは全大腸癌患者の1%未満12)であると推定されているが,正確な頻度は不明である。
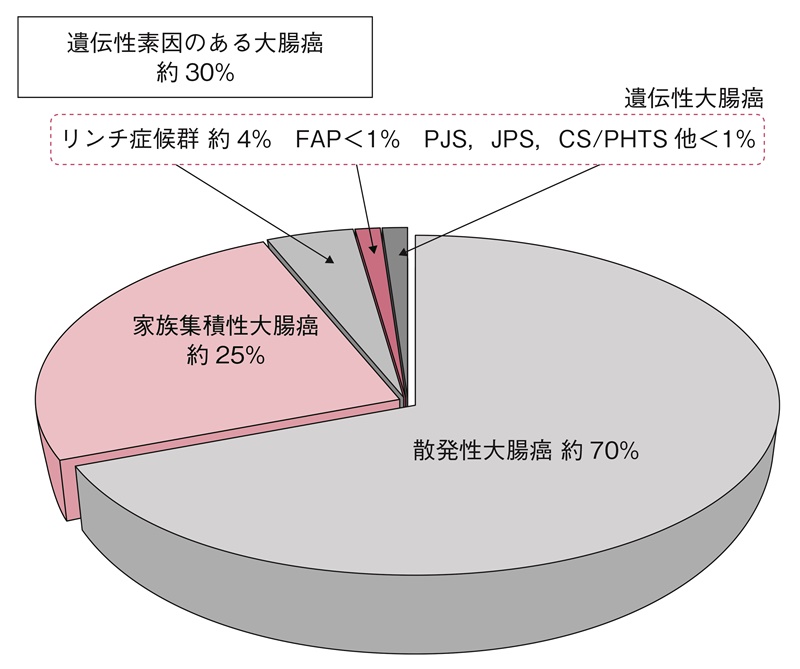
図1 全大腸癌における遺伝性素因のある大腸癌の割合
PJS:Peutz-Jeghers症候群,JPS:若年性ポリポーシス症候群,CS/PHTS:Cowden症候群/PTEN過誤腫症候群
〔主な疾患〕- 遺伝性大腸癌の代表的な疾患(表3)のうち,FAP,ポリメラーゼ校正関連ポリポーシス(polymerase proofreading–associated polyposis:PPAP),リンチ症候群,Peutz–Jeghers症候群(Peutz–Jeghers syndrome:PJS),若年性ポリポーシス症候群(juvenile polyposis syndrome:JPS),Cowden症候群(Cowden syndrome:CS)/PTEN過誤腫症候群(PTEN hamartoma syndrome:PHTS),Li–Fraumeni症候群(Li–Fraumeni syndrome:LFS)は,常染色体優性遺伝形式である。MUTYH関連ポリポーシス(MUTYH–associated polyposis:MAP),MSH3関連ポリポーシス(MSH3–associated polyposis),NTHL1関連ポリポーシス(NTHL1–associated polyposis)は,常染色体劣性遺伝形式をとる。
- FAPを除くポリポーシスの大腸ポリープ数は,だいたい10個以上100個未満である。また,リンチ症候群やLFSの大腸ポリープ数は,数個以内であることが多い。PJS,JPS,CS/PHTSの大腸ポリープ数は0~数十個程度であり,ポリープの組織型は過誤腫で各疾患に特徴的な形態を示す。
- 大腸癌発症リスクは,疾患により異なるが,FAPは浸透率(サイドメモ1:浸透率)がほぼ100%である。リンチ症候群は,原因となるミスマッチ修復遺伝子によって大腸癌発症リスクが異なり13),女性においては,大腸癌と同程度の頻度で子宮内膜癌も発症する14)。また,リンチ症候群関連腫瘍としてさまざまな部位の腫瘍を発症することが知られているので診療科横断的な連携も重要である。
- リンチ症候群とLFSを除く遺伝性大腸癌については,大腸ポリポーシス以外に胃や十二指腸にもポリープが多発する傾向がある。
- 代表的な遺伝性大腸癌とその原因遺伝子は表3の通りである。これら疾患の原因遺伝子は,APC,TP53,PTEN,SMAD4などのがん抑制遺伝子群と塩基のミスマッチや塩基置換関連の修復遺伝子群に大別される。Knudsonにより提唱されたtwo-hit理論16)に基づいて,原因遺伝子の両アレルに病的バリアントを持つことで本来のタンパクの働きが失われることによりがん化を促すことになる。常染色体優性遺伝形式を呈する遺伝性大腸癌の場合,すでに片方のアレルに生殖細胞系列の病的バリアント(first hit)があり,さらに,もう一方のアレルにLOH(loss of heterozygosity)(サイドメモ1:ヘテロ接合性の消失)や新たな病的バリアント等の後天的な変化が起きると(second hit),散発性の大腸癌よりも早期にがん化しやすいと考えられる(図2)。FAPの場合,two-hit目の体細胞におけるバリアントが大腸の上皮細胞に加わると,異常腺窩巣(aberrant crypt foci:ACF)(サイドメモ1:異常腺窩巣)が発生すると考えられている17)。
- 常染色体劣性遺伝形式の遺伝性大腸癌にMAPやMSH3関連ポリポーシス,NTHL1関連ポリポーシスなどがある。すなわち,これらの疾患は,原因遺伝子に生殖細胞系列の病的バリアントを有する保因者である両親からそれぞれの病的バリアントを有するアレルを受け継いだ時に発症する。
- FAPの原因遺伝子であるAPC遺伝子の発見は,adenoma-carcinoma sequenceという散発性大腸癌の主たる発がん機構として提唱された18)。すなわち,遺伝性大腸癌においても,原因遺伝子に加えて,染色体不安定性(chromosomal instability:CIN)(サイドメモ1:染色体不安定性)やMSIなどにより複数の遺伝子異常が蓄積して起こる多段階発がんである(図3)。
- FAPにおける腫瘍では,生殖細胞系列におけるAPC遺伝子の片側アレルの異常に加え,体細胞レベルでの対側アレルに機能喪失型の変化が起こる。
- FAPでは,APCタンパクの機能異常により細胞内に蓄積したβカテニンの細胞質内から核内への移行が増加し,TCF4と複合体を形成した結果,がん遺伝子などの転写が促進され,細胞増殖する。
- FAPにおいて,ACFから腺腫を経て大腸癌が発生するためには,KRAS遺伝子やTP53遺伝子などの発がんに関連する遺伝子に変化が加わると考えられている18)。
- リンチ症候群では,ミスマッチ修復遺伝子の片側のアレルに生殖細胞系列の病的バリアントを有しており,後天的に対側アレルに機能喪失の変化が加わることでミスマッチ機構が損なわれる。その結果,ゲノム内の単純な反復配列であるマイクロサテライト領域に反復回数の異常(不安定性)が好発するようになる。腫瘍抑制(TGFBR2など),細胞増殖,DNA修復(MSH3,MSH6など)やアポトーシス(BAXなど)などに関わる遺伝子産物(タンパク)をコードする領域には反復配列が含まれており,これらの領域に変化が起こりやすい。
- リンチ症候群の原因遺伝子の一つであるMLH1遺伝子は,日本人の場合,全大腸癌の約6%19)で高度メチル化により失活しており,MSI-Hを示す散発性大腸癌(各論Ⅲ 2-2):鑑別を要する疾患参照)の主たる原因である。
表3 代表的な遺伝性大腸癌の遺伝学的および臨床的特徴
| 疾患 | 遺伝形式 | 原因遺伝子 | 頻度 | 大腸ポリープ数 | ポリープの組織型 | 大腸癌発症リスク | 大腸以外のポリポーシス | 大腸癌以外の腫瘍性病変 |
|---|---|---|---|---|---|---|---|---|
| 家族性大腸腺腫症 | 常染色体優性 | APC(5q22.2) | 1/20,000~1/10,000 | 通常100個以上(attenuated typeでは100個未満) | 腺腫 | 40歳代中頃で約50%,60歳以上でほぼ100% | 胃,十二指腸 | 胃癌,十二指腸(乳頭)癌,デスモイド腫瘍,骨腫,甲状腺乳頭癌,脳腫瘍,肝芽腫など |
| MUTYH関連ポリポーシス | 常染色体劣性 | MUTYH(1p34.1) | 不明 | 10~100個 | 腺腫(鋸歯状腺腫の合併) | 60歳まで43~100% | 十二指腸 | 十二指腸(乳頭)癌,甲状腺癌,皮脂腺腫瘍など |
| ポリメラーゼ校正関連ポリポーシス | 常染色体優性 | POLE(12q24.33) POLD1(19q13.33) |
不明 | 0~70個 | 腺腫 | 平均35~40歳 | 十二指腸 | (POLE)十二指腸癌,脳腫瘍(POLD1)子宮内膜癌,乳癌,脳腫瘍 |
| リンチ症候群 | 常染色体優性 | MLH1(3p22.2) MSH2(2p21-p16.3) MSH6(2p16.3) PMS2(7p22.1) EPCAM(2p21) |
全大腸癌における2~4% | 通常数個以内 | 腺腫 | 40歳代中頃(原因遺伝子で異なる) | なし | 子宮内膜癌,胃癌,卵巣癌,小腸癌,胆道癌,膵癌,腎盂・尿管癌,脳腫瘍,皮脂腺腫瘍など |
| Peutz-Jeghers症候群 | 常染色体優性 | STK11(19q13.3) | 1/280,000~1/8,300 | 0~数十個 | 過誤腫 | 生涯リスク39% | 胃,小腸 | 胃癌,小腸癌,膵癌,乳癌,子宮頸部腺癌など |
| 若年性ポリポーシス症候群 | 常染色体優性 | SMAD4(18q21.2) BMPR1A(10q23.2) |
1/100,000~1/16,000 | 0~数十個 | 過誤腫 | 生涯リスク39~68% | 胃,小腸 | 胃癌,小腸癌,膵癌など |
| Cowden症候群/PTEN過誤腫症候群 | 常染色体優性 | PTEN(10q23.31) | 1/200,000 | 0~数十個 | 過誤腫,神経節腫,腺腫,過形成ポリープ | 生涯リスク9~16% | 食道,胃,小腸 | 乳癌,子宮内膜癌,甲状乳頭癌・濾胞癌,腎癌など |
| Li-Fraumeni症候群 | 常染色体優性 | TP53(17p13.1) | 不明 | 数個以内 | 腺腫 | 詳細不明 | なし | 骨・軟部肉腫,副腎皮質腫瘍,脳腫瘍,白血病,乳癌など |
| MSH3関連ポリポーシス | 常染色体劣性 | MSH3(5q14.1) | 不明 | 数十個 | 腺腫 | 詳細不明 | 十二指腸 | 甲状腺腫,乳管内乳頭腫,胃癌,脳腫瘍など |
| NTHL1関連ポリポーシス | 常染色体劣性 | NTHL1(16p13.3) | 不明 | 数十個 | 腺腫 | 詳細不明 | なし | 乳癌,子宮内膜癌,膀胱癌,頭頸部癌,皮膚癌など |
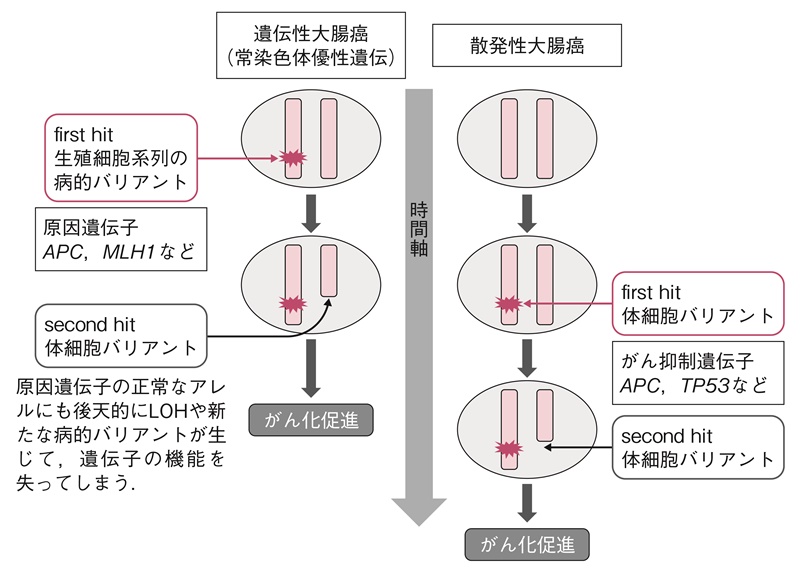
図2 Knudsonのがん抑制遺伝子のtwo-hit説によるがん化のメカニズム
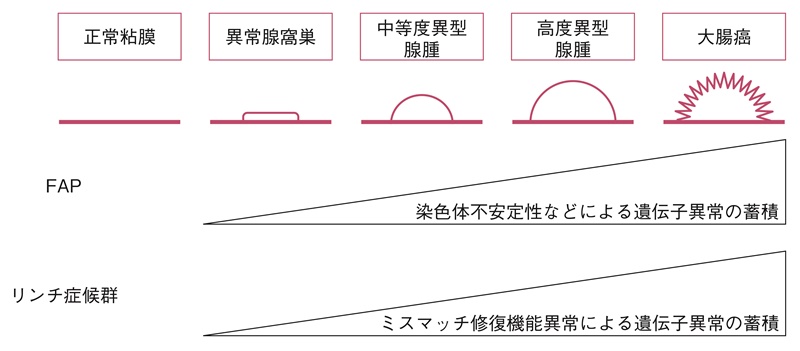
図3 FAPとリンチ症候群の代表的ながん化のメカニズム
サイドメモ1
■バリアント
「バリアント(variant)」とは,遺伝情報の多様性を意味する用語で,主にDNAの塩基配列において,参照配列と異なる塩基配列を指す。日本語訳として「多様体」が用いられることもあるが,「バリアント」のまま用いられることが多い。同様な言葉として「変異」があるが,生物学的意義を持たせた表現として使う場合とそうでない場合があるなど用い方に混乱がある。そのため,「変異(mutation)」という言葉はなるべく用いず「バリアント」を用い,生物学的意義や臨床的意義の評価を付加する場合は,pathogenic(病的)やbenign(病的でない),uncertain significance(意義不明)などの修飾語をつけて表現する。
■生殖細胞系列バリアントと体細胞バリアント
精子あるいは卵子を経由して受け継がれるDNAの塩基配列変化を生殖細胞系列バリアントという。受精卵の時点でその変化は存在するため,全身のすべての細胞に同じ変化が存在する。それに対して,身体を構成する生殖細胞以外の細胞(体細胞)に新たに生じた塩基配列の変化を体細胞バリアントという。
■遺伝学的検査
「遺伝子検査」という用語は,「体細胞の遺伝子検査」なのか「生殖細胞系列の遺伝子検査」なのか区別がつかないため,前者を「体細胞遺伝子検査」,後者を「遺伝学的検査」と呼ぶことが日本臨床検査標準協議会の「遺伝子関連検査標準化専門委員会」から提言された。また,これらの呼称は,遺伝医学関連学会などにより作成された日本医学会の「医療における遺伝学的検査・診断に関するガイドライン」(https://jams.med.or.jp/guideline/genetics-diagnosis.html)でも分類・定義されている。
■浸透率
遺伝性疾患における原因遺伝子の遺伝型(Genotype)の保有者において病気が発症する確率である。100%の確立で発症する場合を完全浸透という。
■染色体不安定性/chromosomal instability:CIN
CINとは,がん細胞などで見られる染色体の数の異常や構造異常(欠失,重複,転座など)のこと。腫瘍化の原因になると考えられている。
■ヘテロ接合性の消失/loss of heterozygosity:LOH
両親から各々受け継がれた遺伝情報のうち,相同な領域において異なる塩基配列が存在する場合をヘテロ接合性(heterozygosity)という。FAPの場合,正常細胞ではAPC遺伝子の片側にのみ病的なバリアントが存在しており,もう一方のAPC遺伝子は正常(野生型)である。この状態がヘテロ接合性であるが,がん化の過程で野生型のAPC遺伝子が欠失により失われることをLOHという。
■異常腺窩巣/aberrant crypt foci:ACF
ACFは,内視鏡の通常観察では正常粘膜と区別することはできないが,拡大視観察ではメチレンブルーに濃染する異常腺管の集合として確認できる。ACFの一部は腺腫や癌の前駆病変と考えられている。2 診 断
- 大腸癌患者の中から遺伝性大腸癌が疑われる患者をスクリーニングする。ポリポーシスの場合,大腸内視鏡検査時のポリープの生検による組織診断で腺腫性ポリポーシスと過誤腫性ポリポーシスかの情報を得ることにより疾患を絞り込みやすくなる。
- ポリポーシス症候群以外の遺伝性大腸癌(リンチ症候群やLFS)では,大腸ポリープの数が少ないため散発性大腸癌と区別がつかない。そこで,病歴や家族歴,切除標本の病理組織診断など遺伝性大腸癌を疑う情報収集に努めることが重要である。
- リンチ症候群の確定診断には遺伝学的検査が必要であるが,現在のところ保険診療では行えない。遺伝性大腸癌が確定している,あるいは疑われる場合は,患者本人のほかに家族(血縁者),特に第1度近親者(親,子,兄弟姉妹)に遺伝カウンセリングを行った上で,実施について検討する。
1)診断の流れ
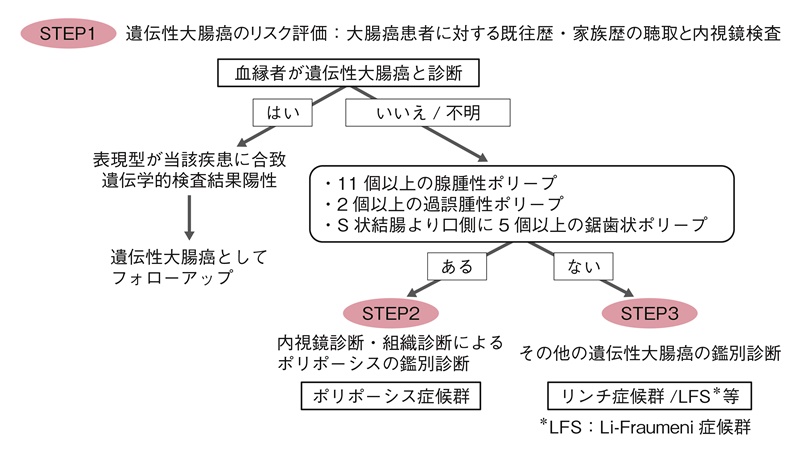
図4 遺伝性大腸癌のリスク評価
STEP1
〔遺伝性大腸癌のリスク評価:問診と内視鏡検査〕- 日常臨床において,遺伝性の大腸癌であるかどうかの手掛かりは,本人の病歴として①若年発症,②同時性・異時性の大腸癌,③大腸癌以外の悪性腫瘍の既往など遺伝性大腸癌の特徴を有しているか,さらに父方,母方の両方の家系について第3度近親者(少なくとも第2度近親者)まで家族歴の聴取をすることが基本である。核家族化がすすみ,第2度近親者の情報さえも得られないこともあるが,父方と母方を区別して遺伝形式を評価するために必要な情報を収集し,家系図を作成することを推奨する(付録Ⅰ:家系図の書き方・読み方の原則)。
- ポリポーシスではない遺伝性大腸癌,たとえばリンチ症候群は,リンチ症候群関連腫瘍の既往歴や家族歴の聴取が不可欠である。この場合はSTEP3へすすむ。
STEP2
〔大腸ポリポーシスの鑑別診断(図5)〕- 大腸内視鏡検査で大腸ポリープの生検による組織診断で腺腫性ポリポーシス,過誤腫性ポリポーシス,鋸歯状ポリポーシスの疾患群に分ける。
- 腺腫性ポリポーシスのうち,ポリープの数が100個以上の場合は,FAPを第一に考える。腺腫性ポリープの数が,11個以上100個未満の場合は,頻度順に①attenuated FAP(AFAP),②MAP,③PPAPなどが候補になる。これらのポリポーシスは,内視鏡所見での鑑別はできないので,確定診断には家族歴などを聴取した後,遺伝学的検査(サイドメモ1:遺伝学的検査)を行う必要がある。
- 腺腫性ポリポーシスの場合,常染色体優性遺伝形式の家族歴かどうかは,診断のための有用な情報となる。さらに,胃底腺ポリポーシスや十二指腸腺腫,外骨腫,先天性網膜上皮肥大などの随伴症状の有無が参考になる。一方,両親に疾患が観察されない場合は,MAP等の常染色体劣性遺伝疾患や体細胞APCモザイク,新生発端者などの可能性について検討する。
- 過誤腫性ポリポーシス疾患群の消化管ポリープの数は,PJSとJPSでは数個から数十個,CS/PHTSで大半が50個以上である。PJSとJPSのポリープは,組織学的に特徴的な形態を呈しており,ポリープ数とともに臨床診断項目に含まれている13)。各疾患とも消化管ポリポーシス以外のさまざまな病態を併せ持っているので,各診断基準に沿って診断できる場合がある。
STEP3
〔その他の遺伝性大腸癌の鑑別診断〕- リンチ症候群やLFSを拾い上げる手段としては,問診により世代間に渡って大腸癌やそのほかのリンチ症候群関連腫瘍,LFSに高い頻度で認められる乳癌,骨軟部腫瘍や脳腫瘍などに罹患された血縁者が家系内にいないかどうか調べることは重要である。
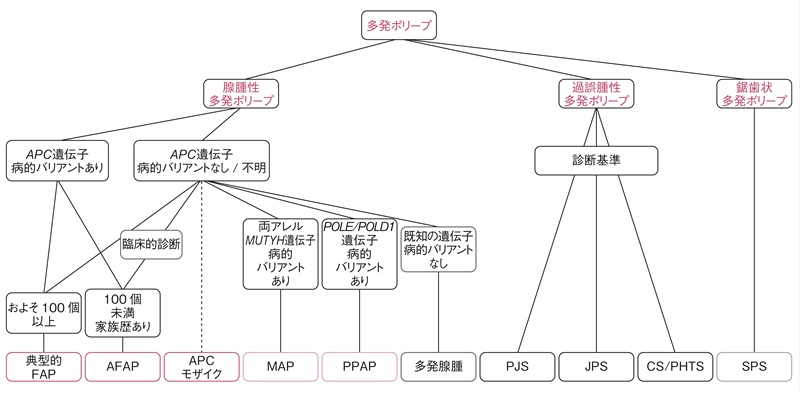
図5 遺伝性大腸ポリポーシス 診断のフローチャート
FAP:familial adenomatous polyposis,AFAP:attenuated FAP,MAP:MUTYH関連ポリポーシス,PPAP:polymerase proofreading-associated polyposis,PJS:Peutz-Jeghers症候群,JPS:若年性ポリポーシス症候群,CS/PHTS:Cowden症候群/PTEN過誤腫症候群,SPS:serrated polyposis症候群
2)遺伝学的検査
〔臨床的意義〕- 腺腫性ポリポーシスのうち,典型的なFAPは臨床症状から診断できるものの,その他の疾患は遺伝学的検査により,原因遺伝子の病的バリアントを同定することで確定診断できる。
- リンチ症候群の確定診断には,遺伝学的検査で原因遺伝子の病的バリアントを同定する必要がある。
- 遺伝性大腸癌の発端者に原因遺伝子の病的バリアントが同定されれば,血縁者の診断も可能になる。
- 遺伝子解析に必要な量の採血をする(通常2~10 mL程度)。
- 遺伝学的検査では,疾患の原因遺伝子のタンパクをコードするエクソンとエクソン-イントロン境界領域の塩基配列を解析する。エクソンレベルの比較的大きな欠失や重複は,multiplex ligation-dependent probe amplification(MLPA)法にて解析する。
- 発端者の遺伝子異常が同定されている場合,血縁者の遺伝学的検査は,バリアントの部位のみ(シングルサイト)の解析でもよい。
- 遺伝性大腸癌の遺伝学的検査には,①疑われる遺伝性腫瘍の推定される原因遺伝子一種類のみを対象とした検査,②疑われる遺伝性腫瘍の推定される複数の原因遺伝子を対象とした検査,③鑑別診断に必要な疾患を含めた原因遺伝子がセットになったmulti-gene panel検査,④遺伝性腫瘍の原因遺伝子を網羅的に含むmulti-gene panel検査,⑤全エクソン,全ゲノム検査がある(表4)。
表4 遺伝学的検査の種類
例)FAPの場合 APC
②疑われる遺伝性腫瘍の推定される複数の原因遺伝子を対象とした検査
例)リンチ症候群の場合 MLH1/MSH2/MSH6/PMS2/EPCAM
③鑑別診断に必要な疾患の原因遺伝子がセットになったmulti-gene panel検査
④遺伝性腫瘍の原因遺伝子を網羅的に含むmulti-gene panel検査
⑤全エクソン,全ゲノム検査
- ①の例として,FAPが疑われた場合,原因遺伝子であるAPCのみの遺伝学的検査を行う。
- ②の例として,リンチ症候群が疑われた場合,MLH1,MSH2,MSH6,PMS2に加えて,MSH2遺伝子の上流に隣接してMSH2遺伝子のプロモーター領域に異常メチル化を引き起こしMSH2タンパクの発現消失に関与するEPCAM遺伝子の欠失の解析も含まれている遺伝子セットの検査が相当する。
- ③の例として,数十個の多発大腸ポリープに対し,考えられる遺伝性腫瘍が複数あり,臨床的所見での鑑別が困難な場合があげられる。その場合は,それら疾患の原因遺伝子が含まれている遺伝子セットを調べることで,一度に効率よく原因を特定できる可能性がある。それでも原因が特定できなかった場合,④の遺伝性腫瘍の原因遺伝子を網羅的に含むmulti-gene panel検査や⑤全エクソン,全ゲノム検査を行うことがある。
- 遺伝性大腸癌の遺伝学的検査は,現在のところ保険診療外であるが,自費診療での外注,あるいは一部の施設では研究レベルでも実施することが可能である。
- 標準治療がない固形がん(原発不明がんや希少がんなど),または標準治療が終了となった固形がん(終了が見込まれる者を含む)患者を対象としたがん遺伝子パネル検査が2019年5月に保険適用となった。これらのがん遺伝子パネル検査(表5)は,本来は有効な薬物の探索を目的とした検査であるが,遺伝性大腸癌の原因遺伝子も含まれているため,遺伝性大腸癌が診断される可能性がある。
表5 がん遺伝子パネル検査に含まれる遺伝性大腸癌の原因遺伝子
- OncoGuide™ NCCオンコパネルシステム
APC, MLH1, MSH2, POLD1, POLE, PTEN, SMAD4, STK11, TP53 - FoundationOne® CDxがんゲノムプロファイル
APC, MLH1, MSH2, MSH3, MSH6, MUTYH, PMS2, POLD1, POLE, PTEN, SMAD4, STK11, TP53
- 検出されたバリアントの臨床的意義については,ClinVar(https://www.ncbi.nlm.nih.gov/clinvar/)やInSiGHT(https://www.insight-group.org/variants/databases/)のクラス分類(表6)等で評価する。
表6 遺伝子バリアントのクラス分類
ClinVar
|
InSiGHT
|
―pathogenicまたはlikely pathogenicに相当
- 遺伝性疾患として医学的管理を行う。ただし,病的バリアントを有する未発症者が,FAPのような浸透率がほぼ100%である疾患を除いては,生涯に必ずがんを発症するとは限らないことも理解してもらう。
―uncertain significanceに相当
- 疾患への影響が分からない遺伝子変化のことをVUS(variants of uncertain significant)として報告される。例えば,塩基配列が一つ変化してもアミノ酸合成には影響しないサイレントバリアントや,アミノ酸の置換が起きるミスセンスバリアントが疾患発症に影響するのか判断できない場合である。この場合,このバリアントの意義が証明されるまで,次項の「遺伝子異常が検出されない」と同様に対応することがすすめられる。
―benignまたはlikely benignを含む
- 〔家系内で遺伝子異常が確定している場合〕
- 遺伝学的検査で確定診断されている家系において,同じ遺伝子変化が認められなかった場合は,家系で認められた遺伝性疾患ではないと判断する。その場合でも,一般集団の発がんリスクがあることは理解してもらう。
- 〔家系内の確定診断がされていない場合〕
- 遺伝学的検査に用いられた方法によっては検出できないバリアントを有しているか,あるいは未知の原因遺伝子の異常によることなども考えられ,慎重に対応する。例えば,臨床的にポリープ数が100~1000個ほどあるFAPでも,APC遺伝子の病的バリアントの検出率は60%程度20)と100%には至らないことから,技術的な問題で未知のバリアントが検出されていないことやAPC以外の遺伝子の異常の可能性も考えなければならない。臨床的に遺伝性疾患と考えられる場合,遺伝学的検査で病的バリアントが検出されなくても,実地臨床では遺伝性疾患と同様に対応していくことが望ましい。
3)遺伝カウンセリング
〔概要〕- 遺伝カウンセリングでは,受容的態度,非指示的対応,共感的理解のカウンセリングの3原則を守る必要がある。遺伝カウンセリングでは正確な遺伝医学の知識をわかりやすく伝えることにより,遺伝的問題で悩む患者家族の不安を取り除く。相談者の考え方,感受性,事前の知識,理解力,不安の大きさ,医療に対する信頼感が個々で異なることに注意する。
- 遺伝性大腸癌は,散発性大腸癌とは異なり,さまざまな随伴病変を有するので専門的な医学的管理のもと長期のサーベイランスを要する。したがって,遺伝性大腸癌の特徴を持った患者には,遺伝カウンセリングや遺伝学的検査の実施体制が整備された専門施設への紹介を検討する。
- 遺伝学的検査を施行する前にその臨床的意義について説明し,検査による利点と問題点(表7)について理解していることが必須である。
表7 遺伝学的検査の利点と問題点
| 利点 | 問題点 |
|---|---|
| ・疾患の確定診断が得られる。 ・少量の血液検査で調べられる。 ・家族歴の有無に関係なく診断できる。 ・罹患する可能性のある疾患に対するサーベイランスが行える。 ・血縁者が罹患しているか血液検査で診断可能である。 |
・遺伝学的検査には限界があり,検出できないこともある。 ・診断が得られても,必ずしも発症を予測できないこと。 ・自費診療であること。 |
- 遺伝性大腸癌の原因遺伝子の遺伝学的検査は,本邦では保険収載されておらず自費診療となるため,費用の負担がかかる一方で,必ず病的バリアントが検出できるわけではないという検査の限界についても説明する必要がある。
- 遺伝学的検査は,医学的,倫理的,経済的,技術的なさまざまな観点でクライアントの負担にならないように配慮しながら,文章による検査の説明と同意書を作成し,インフォームド・コンセントを得た上で実施する。遺伝学的検査を受ける前後だけではなく,必要に応じて遺伝カウンセリングを継続する。
- 遺伝学的検査の結果開示時には,家族の同席について希望の有無を確認する。同席を希望しない場合,個別に時間と場所を確保する。
- 患者本人の他に,家族(血縁者)にも遺伝カウンセリングを行うことが望ましい。
- 第1度近親者(親,子,兄弟姉妹)には疾患について十分な説明を行い,同意を得た上でリスク評価に応じた関連腫瘍のサーベイランスを行う。
- 遺伝学的検査の実施に際し,日本医学会の「医療における遺伝学的検査・診断に関するガイドライン」(https://jams.med.or.jp/guideline/genetics-diagnosis.html),日本遺伝性腫瘍学会の「家族性腫瘍における遺伝学的検査の研究とこれを応用した診療に関する指針(2019年版)」(http://jsht.umin.jp/information/opinion/download/guideline2019040101.pdf),文部科学省,厚生労働省,経済産業省3省取りまとめの「ヒトゲノム・遺伝子解析研究に関する倫理指針」(平成13年4月1日施行,平成29年2月28日一部改正)(https://www.mhlw.go.jp/file/06-Seisakujouhou-10600000-Daijinkanboukouseikagakuka/0000153405.pdf)などを遵守する。また,被検者のプライバシーに配慮し,記録の保管は慎重に対処する。
- クライアントが遺伝性大腸癌について正確に理解するために,がんと遺伝に関するさまざまな情報を提供する必要がある(表8)。また,遺伝学的検査を行う際に結果によって本人や家族にもたらす心理的影響や社会的差別への配慮などにも留意する。
- 遺伝に関連して発症する腫瘍の発症時期をもとに,遺伝学的検査の適切な時期を検討する。例えば,リンチ症候群の関連腫瘍の発症は一般に成年期以降であるので,遺伝学的検査の時期も原則的に成年期以降になる。遺伝学的検査で確定診断がついている血縁者,および遺伝学的検査を行っていない血縁者は,リンチ症候群としての関連腫瘍のサーベイランスを行う。
表8 カウンセリング時の情報提供
- ①該当する遺伝性疾患について
- 遺伝性疾患が疑われる理由―発症要因(環境や遺伝)
- 疾患の原因遺伝子
- 遺伝形式―常染色体優性(あるいは劣性)遺伝,血縁者が病的バリアントをもつ確率
- 遺伝性腫瘍の特徴―がんの浸透率
- がんに対する対策―予防,早期発見,治療
- ②遺伝学的検査について
- 検査の目的,方法,検査精度や検出率,検査の限界・不確実性,費用
- 期待される利益―確定による不確実性からの不安の解消,発症リスクの予測,予防的治療,血縁者の発症診断にも有用であること
- 本人や家族にもたらす心理的影響,子供に遺伝する可能性
- 検査を受けなかった場合の今後の対策や選択肢
- 患者本人のほかに家族(血縁者)にも遺伝カウンセリングを行うことが望ましい。
- 第1度近親者(親,子,兄弟姉妹)には疾患について十分な説明を行い,同意を得た上で大腸を中心とする消化管サーベイランスを行う。
- サーベイランスの必要性,遺伝子診断の意義についての情報を提供する。
- 大腸癌の発症リスクは疾患によって,また原因遺伝子のバリアントの種類等によって大きく異なる。
- 遺伝性大腸癌は,長期間のサーベイランスが必要である。
Ⅱ.家族性大腸腺腫症(familial adenomatous polyposis:FAP)
1 概 要
- 家族性大腸腺腫症(familial adenomatous polyposis:FAP)は,APC遺伝子の生殖細胞系列バリアントを原因とし,大腸腺腫の多発を主徴とする常染色体優性遺伝性の疾患である。
- 放置すると患者のほぼ100%に大腸癌が発生する。
- 大腸癌以外にも,消化管その他の臓器にさまざまな腫瘍性および非腫瘍性の随伴病変が発生する。
- 大腸癌の発生は10歳代での報告もあるが,40歳代でほぼ50%,放置すれば60歳頃にはほぼ100%に達する21)(資料Ⅰ:家族性大腸腺腫症,資料図1:大腸癌の累積発生率)。
- FAPの死因22)の第1位は大腸癌で,その割合は1980年代までは約80%であったが,90年代以降は約60%と減少傾向にある。
- 主な大腸外随伴病変(表9)のうち,デスモイド腫瘍,十二指腸癌は大腸癌以外のFAPの主要な死因であり,その頻度はそれぞれ約10%,約6%である22)。
- 第5番染色体上のAPC遺伝子(5q22.2)
- 常染色体優性遺伝
- 全人口における頻度は,欧米では1:20,000から1:10,000,わが国では1:17,400と推定されている23)。全大腸癌患者のうち,1%未満がFAP患者と推定されている12)。
2 診 断
1)診断の流れ(各論Ⅰ 図5:遺伝性大腸ポリポーシス 診断のフローチャート参照)
- FAPの診断は臨床的または遺伝子診断により行われる。
【臨床的診断】
(1)または(2)に合致する場合はFAPと診断する。
(1)大腸にほぼ100個以上の腺腫を有する。家族歴の有無は問わない。
(2)腺腫の数は100個に達しないがFAPの家族歴を有する。
【遺伝子診断】(サイドメモ1)
APC遺伝子の生殖細胞系列バリアントを有する場合はFAPと診断する。
- 大腸におよそ100個以上の腺腫がある場合でもFAPと診断できない例外(劣性遺伝形式のMUTYH関連ポリポーシス)がある。したがって優性遺伝に矛盾しない家族歴はFAPの補助診断としてきわめて有用である。
- 大腸外随伴病変は大腸腺腫の個数にかかわらずFAPの補助診断として有用である。
- 臨床的にFAPと診断されても,その20~40%にはAPC遺伝子バリアントが検出されない20,24)。
- 患者が自身の診療や血縁者の診断のために遺伝学的検査を希望する場合,あるいはattenuated FAP(AFAP)とMUTYH関連ポリポーシスやポリメラーゼ校正関連ポリポーシス(polymerase proofreading-associated polyposis:PPAP)との鑑別が必要な場合,APC遺伝子の遺伝学的検査を考慮する。APC遺伝子の遺伝学的検査は検査会社で実施可能である(保険収載されていない)(サイドメモ2:APC関連ポリポーシス)。(CQ1)
サイドメモ2
■APC関連ポリポーシス/APC-associated polyposis conditions
APC遺伝子の生殖細胞系列の病的バリアントを原因とするポリポーシスをAPC関連ポリポーシスと呼称することがある25)。APC関連ポリポーシスには,APC遺伝子のプロモーター1B領域の病的バリアントを原因とするgastric adenocarcinoma and proximal polyposis of the stomach(GAPPS)も含まれる。
■Gastric adenocarcinoma and proximal polyposis of the stomach(GAPPS)
GAPPSは,APC遺伝子のプロモーター1B領域における生殖細胞系列の病的バリアントを原因とし,胃底腺ポリポーシスを主徴とする26)。大腸においてAPC遺伝子の発現を主に制御しているプロモーター1A領域は,胃ではメチル化により不活化されている。そのため,プロモーター1B領域の異常は,胃におけるAPC遺伝子の発現を抑制する25)。
2)腺腫密度による分類
- 腺腫密度により,密生型FAP,非密生型FAP,attenuated FAPに分類されることがある。密生型FAPと非密生型FAPをあわせて,典型的(古典的)FAPとも呼称される。
- 腺腫密度はAPC遺伝子の生殖細胞系列バリアントの部位や大腸癌発生のリスクと関連する。
- 密生型FAP(severe/profuse/dense FAP):肉眼的に正常粘膜が観察できないほど腺腫を発生する場合(図6)(サイドメモ3:密生型と非密生型の境界)。ただし,大腸の部位によって腺腫密度が異なることもしばしば経験する。
- 非密生型FAP(sparse FAP):正常粘膜を背景に腺腫が多発し,腺腫数がおよそ100個以上の場合(図7)。
- AFAP(attenuated FAP)注1:腺腫数が,およそ10個以上100個未満の場合。
- 密生型FAPではAPC遺伝子のcodon 1250~1464(特にcodon 1309)27,28),AFAPでは,APC遺伝子の5’側や3’側の領域のほかに選択的スプライシング領域(バリアントにより特定のexonが転写時に読み飛ばされる領域)に生殖細胞系列バリアントが認められることが多い29)。
- 大腸癌研究会の多施設共同研究によると,密生型FAPではその他のFAPと比べて腺腫発生の年齢やがん化の年齢も早く,密生型で40歳,非密生型で47歳,attenuated型で55歳になると半数に大腸癌の発生がみられた。
注1 attenuated FAP:軽症型FAP,希薄型FAP,散発型FAPなど定訳はない。
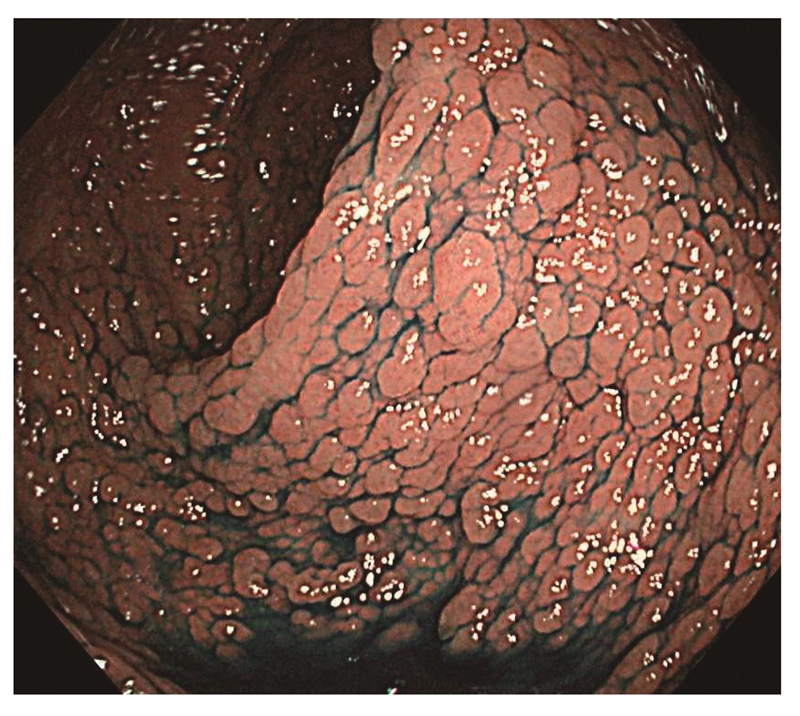
図6 密生型FAP
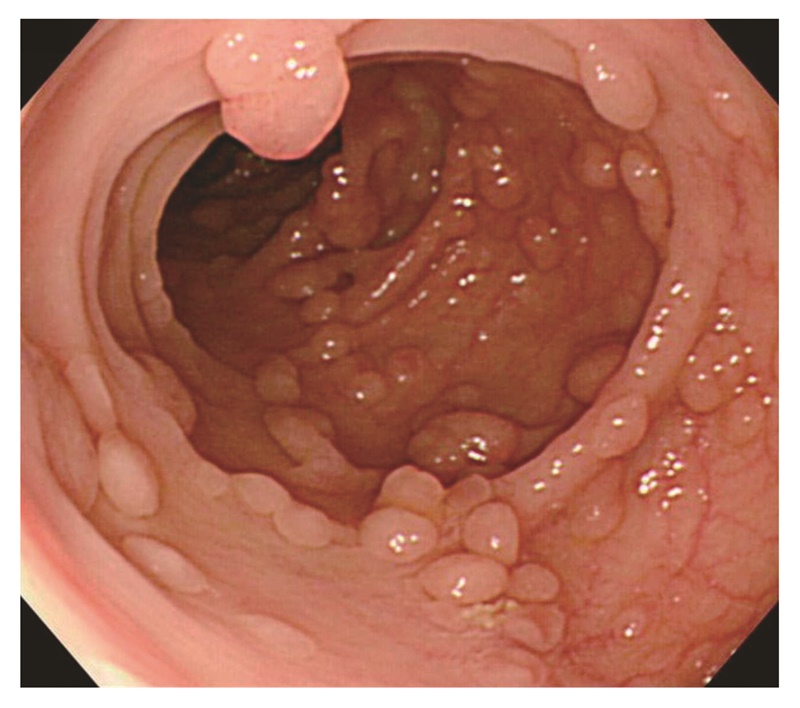
図7 非密生型FAP
サイドメモ3
■密生型と非密生型の境界
大腸腺腫の個数によって密生型>1,000個(または2,000個),非密生型100~1,000個(または2,000個)に分類されることがある。これらを典型的FAPとし,腺腫数の少ないAFAP(attenuated FAP)は10~99個とする報告が多い。密生型と非密生型を厳密に区別する臨床的意義は乏しい。
3)鑑別を要する疾患・病態
体細胞APCモザイク:
APC遺伝子の体細胞バリアントが個体発生の過程で起こった場合,APC遺伝子にバリアントがある細胞とない細胞から構成されるモザイク状態が生じる。大腸の粘膜細胞に分化する細胞にこの異常が起きると,FAP同様大腸腺腫が多発する。APC遺伝子バリアントが明らかになったFAP患者の1.6~4%にAPCモザイクが認められ,家族歴のないFAPの11~20%が体細胞APCモザイクであったと報告されている30,31)。近年,次世代シークエンサーを用いて生殖細胞系列における低頻度の病的バリアントを検出することが可能になり,通常の方法では病的バリアントが認められなかったFAP患者の25~50%に低頻度の病的バリアントが認められる32,33)。臨床的にはFAPとして対応する。また,APC遺伝子バリアントが生殖細胞の一部に存在する(性腺モザイク)場合は,次世代にも遺伝する可能性がある。
MUTYH関連ポリポーシス(MUTYH-associated polyposis:MAP):
MAPは,塩基除去修復遺伝子の一つであるMUTYH遺伝子の両アレルにおける生殖細胞系列の病的バリアントを原因とする常染色体劣性遺伝性疾患34)である。大腸に10~100個の腺腫を認めるのが特徴であるが,100~1,000個の場合もある35)。日本人におけるMUTYH遺伝子バリアントの頻度や全大腸癌に占める割合は不明である。大腸癌の浸透率(遺伝子バリアントを有する症例中で大腸癌を発症する人の割合)は60歳までで43~100%である36)。MAPの患者ではFAPと同様の随伴病変が報告されている。本邦では本疾患の報告は少なく,不明な点も多い。治療はAFAPに準じて行われる。
ポリメラーゼ校正関連ポリポーシス(polymerase proofreading-associated polyposis:PPAP):
PPAPは,DNA複製の際のエラーを修復する機能(校正機能)を持つPOLE遺伝子やPOLD1遺伝子の生殖細胞系列の病的バリアントを原因とする常染色体優性遺伝性疾患である37)。大腸の腺腫の数は数十個であることが多いが,腺腫を合併しない症例も報告されている。PPAPの大腸外病変として,POLE遺伝子が原因の場合には,十二指腸腺腫・癌や脳腫瘍38)を,POLD1遺伝子が原因の場合には,子宮内膜癌や,乳癌,脳腫瘍を併発するとの報告39)がある。PPAPに発生する大腸の腫瘍(大腸腺腫・大腸癌)は,通常の大腸癌と組織学的に区別がつかないので,確定診断のためには遺伝学的検査が必要となる。
3 随伴病変
FAPにおける大腸外随伴病変
- 腫瘍性あるいは非腫瘍性の大腸外随伴病変が合併する(表9)。
表9 FAPに随伴する主な腫瘍性病変
| 胃底腺ポリポーシス* | 頭蓋骨腫,顎潜在骨腫,過剰歯,埋没歯 |
| 胃腺腫* | 類上皮腫 |
| 十二指腸腺腫* | 甲状腺癌 |
| 十二指腸乳頭部腺腫* | 先天性網膜色素上皮肥大 |
| 空・回腸腺腫* | 肝芽腫 |
| デスモイド腫瘍 | 副腎腫瘍 脳腫瘍 |
*:癌化の可能性がある
1)胃底腺ポリポーシス・胃腺腫
- 胃底腺ポリポーシス(図8),胃腺腫(図9)(CQ2)は,FAPの補助診断として参考になる。
- FAP患者において,Helicobacter pylori非感染者に胃底腺ポリポーシスが多い傾向がある40)。しかし,FAPの胃底腺ポリープの一部は悪性化する可能性もあるためサーベイランスが必要である。
- FAP患者には,陥凹型や隆起型の胃腺腫が発生する(図9)。
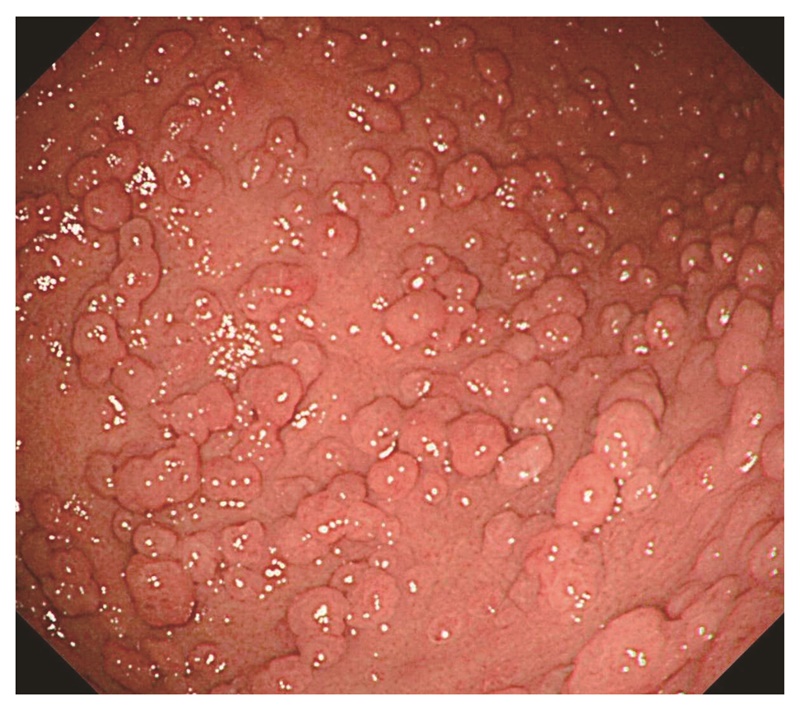
図8 胃底腺ポリポーシス
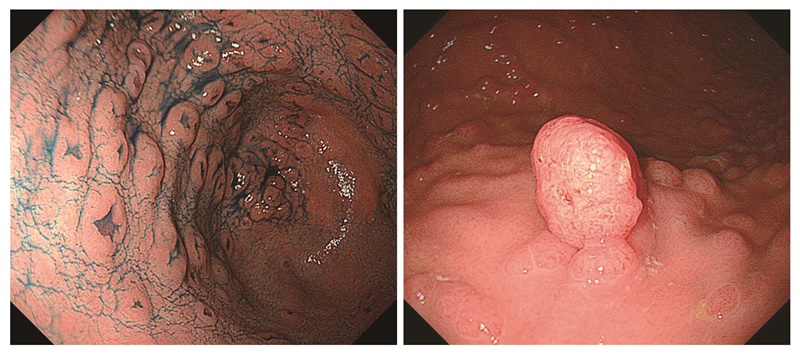
図9 胃腺腫(左:陥凹型,右:隆起型)
2)十二指腸腺腫・癌

図10 十二指腸乳頭部腺腫および十二指腸腺腫
- 十二指腸腺腫はFAP患者の30~90%に認められ41-43),腺腫有病率は40歳以降高くなる42,43)。
- 十二指腸腺腫の発育はきわめて緩徐だが42,44),定期的内視鏡サーベイランス治療が必要である。(CQ2)
- 十二指腸癌は,FAP患者の死因の約3%を占める22,45)。
- FAP患者の十二指腸癌の一般集団に対する相対リスクは250~330.8倍であり46,47),十二指腸癌の累積発生率は57歳で4.5%程度48)と考えられている。
- 大腸癌研究会の多施設共同研究によれば,十二指腸腺腫の50歳までの累積発生率は39.2%で,典型的FAPはAFAPと比較して有意に累積発生率が高かった(42.5% vs. 23.5%)49)。
- 十二指腸腺腫の臨床病理学的分類としてスピゲルマン(Spigelman)の分類がある50)。
- スピゲルマン分類は,内視鏡検査で十二指腸腺腫の個数,最大径を評価し,さらに腺腫の生検組織像(図11)について,細胞構造と異型度を評価する。現在では若干の修正(修正スピゲルマン分類)が加えられている50)(図12)(サイドメモ4:スピゲルマン分類の評価法の変遷)。
- 十二指腸腺腫の診断には,直視内視鏡,斜視内視鏡を用いる。
- 狭帯域光観察(narrow-band imaging:NBI)の使用により十二指腸腺腫の同定数は増えるが,スピゲルマン病期分類には影響を与えなかった51)。
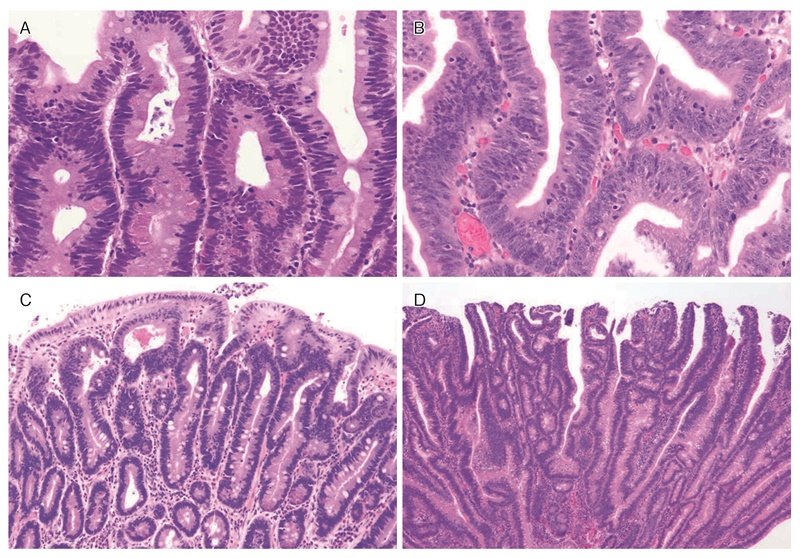
図11 FAPに合併する十二指腸腺腫の組織像
A:低異型度腺腫(low-grade adenoma):腫瘍腺管は比較的整然と配列する。小型紡錘形の核が基底側に配列している。
B:粘膜内癌:腫瘍腺管は不規則さを増し,核の重層化が目立つとともに核小体もしばしば認められる。スピゲルマン分類でhigh-grade adenomaとされる病変には,日本の診断基準で非浸潤性の粘膜内癌に相当する病変が含まれる。
C:管状腺腫(tubular adenoma):単純な腺管状の増殖を示す。
D:管状絨毛腺腫(tubulo-villous adenoma):狭小な間質を伴った絨毛状構造の混在を認める。
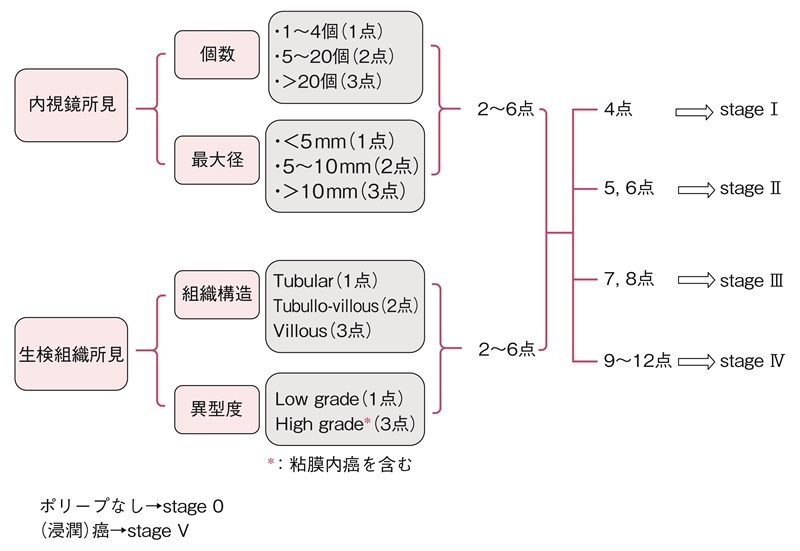
図12 修正スピゲルマン分類による十二指腸腺腫の評価法
- 検査間隔についてのコンセンサスは得られていないが,病期0では4~5年毎,病期Ⅰでは2~5年毎,病期Ⅱでは2~3年毎,病期Ⅲでは6カ月~2年毎に行うことが推奨されている43,48)。
- 修正スピゲルマン病期分類に準じた十二指腸腺腫への対応の目安を図13に示す。
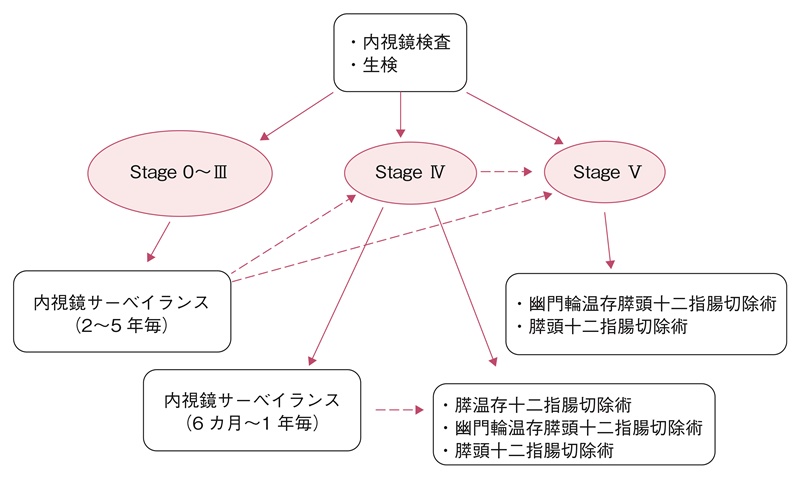
図13 修正スピゲルマン分類に基づいた十二指腸腺腫のサーベイランス・治療方針
- FAP患者の十二指腸病変に対し,内視鏡的治療と経過観察を比較した臨床試験はない。
- 十二指腸腺腫に対する内視鏡的治療にはスネアによる摘除,焼灼,アルゴンプラズマ凝固などがある。
- スピゲルマン病期Ⅰ/Ⅱに分類される腺腫では内視鏡的焼灼が選択されるが,腺腫数が多い場合,内視鏡的あるいは十二指腸切開による切除では不十分な対応となる48)。
- スピゲルマン病期Ⅱ/Ⅲに対する内視鏡的完全摘除は合併症が多く,50~100%の再発率が報告されている41)。
- 病期Ⅳでは7~36%48,52)に癌化が認められるため,手術適応の評価あるいは6~12カ月毎の専門家によるサーベイランスが推奨される。(CQ3)
サイドメモ4
■スピゲルマン分類の評価法の変遷
スピゲルマン分類は1989年に提唱されたFAPに合併する十二指腸腺腫の病期分類である50)。腺腫の個数,最大径,組織構造,異型度について,各々1~3点が振り分けられ,合計点数から病期が決定される。2000年のVienna分類53)によって異型度がmild/moderate/severeの3段階からlow-grade/high-gradeの2段階に変更されたため,low-gradeを1点,high-gradeを3点とする修正病期分類が提唱された54)。最近,NCCN guidelines(Genetic/Familial High-Risk Assessment:Colorectal V.2.2015)ではスピゲルマン分類ないし修正スピゲルマン分類を簡略化した分類が提案されている。この分類では病期0(腺腫なし),病期Ⅰ(1~4個の管状腺腫,1~4 mm大),病期Ⅱ(5~19個の管状腺腫,5~9 mm大),病期Ⅲ(20個以上あるいは1 cm以上の腺腫性病変),病期Ⅳ(腺腫が密生,あるいはhigh-grade adenoma)に分類されている。これらの病期分類を用いたサーベイランスや治療の妥当性について,前向き研究は行われておらず,今後の課題である。
3)デスモイド腫瘍
- デスモイド腫瘍(図14)は,FAPの補助診断として参考になる。
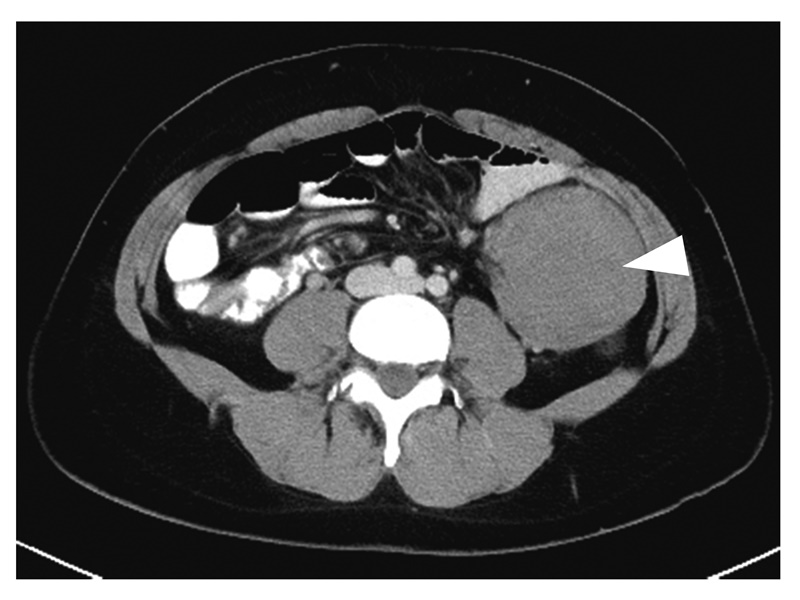
図14 腹腔内デスモイド腫瘍(▽)
- デスモイド腫瘍は線維腫の一種で,転移はしないが浸潤性に発育する傾向がある。
- 腹腔内デスモイド腫瘍がデスモイド腫瘍全体の70%を占め55),大腸切除後(特に2~3年以内,本邦では中央値が26.3カ月)の腹壁・腸間膜あるいは後腹膜に発生することが多い56-58)。
- FAP患者の8~20%に認められる59-62)。
- デスモイド腫瘍は,自然消退ないし安定化することがある63-65)。
- 本邦のデータでも発生率が10~15%で58,66),腹腔内デスモイド腫瘍がデスモイド腫瘍全体の71.8~80%であり,年齢が30歳以下,性別では女性に有意に多く認められ,女性の場合,約2/3が1年以内に発症している58,66)。
- 腹腔内(後腹膜を含む)に発生した場合には,消化管通過障害,穿孔,膿瘍,あるいは尿管閉塞などの原因となり,しばしば治療に難渋する。
- 従来,術式別のデスモイド腫瘍発生リスクには差がないとの報告に対して,本邦からのデータを含む最近の2つの報告では,大腸全摘術(IPAA)の方が部分切除(IRA)よりもデスモイドの発生リスクが高かった58,67)。
- デスモイド腫瘍が発生した場合の死亡率は0~14%と考えられる56,60,62,63,68)。
- これまでにデスモイド腫瘍の発生とAPC遺伝子の病的バリアントの相関性についてはAPCのcodon1444よりも3’側または1445~1580における病的バリアントに相関性を認めるという報告があるが69,70),Churchらの報告ではcodon1399よりも3’側に病的バリアントを持つ症例に高い罹患率を認め,症状が強く致死的な傾向を認めたものの,APCのバリアント部位とデスモイド腫瘍の発生との間に相関性は認めなかった71)。
- 大きな,あるいは発育の早い腹腔内デスモイド腫瘍,あるいは腹壁デスモイド腫瘍には薬物療法を考慮する。(サイドメモ5:デスモイド腫瘍に対する薬物療法)
- 放射線治療は腸管障害をきたす可能性がある上,効果に乏しい72)ので,一般的には推奨されない。
サイドメモ5
■デスモイド腫瘍に対する薬物療法
【NSAIDs・抗エストロゲン薬】
非ステロイド性抗炎症剤(NSAIDs)の一つであるsulindac(300 mg/日)や抗エストロゲン薬のtamoxifen(40~120 mg/日)やraloxifene(120~240 mg/日),toremifene(180 mg/日)の投与ならびにNSAIDsと抗エストロゲン薬の併用についての安全性や有効性に関する報告がある73-75)。Sulindacや抗エストロゲン薬は,腫瘍を縮小させる効果には乏しいものの,腫瘍の増大を抑える効果が報告されているが,効果が認められるまでに平均15カ月かかるとの報告もあり長期的観察を要する75-78)。
【分子標的薬】
最近ではチロシンキナーゼ阻害薬であるimatinibの効果も検討されており,Desurmontら77)は36%の腫瘍縮小あるいは安定化が得られたと報告している。一方,Chughら79)は手術不能のデスモイド腫瘍に対し,1年の無増悪率が66%であるものの,腫瘍縮小は3%と報告しており,現時点ではimatinibの効果は十分明らかとは言えない。
【殺細胞性化学療法】
殺細胞性化学療法(cytotoxic chemotherapy)については,主にdoxorubicin(DOX)とdacarbazine(DTIC)の併用療法において高い奏効率が示されている80)。わが国でもDOX+DTIC療法の有効性が報告されているが毒性も高い点が問題となっている81-83)。DOX+DTIC療法以外では,methotrexate(MTX)とvinblastine(VBL)の有用性が報告されている80)。
【治療法別効果】
Desurmontら77)は各種薬物療法ごとの腹腔内デスモイド腫瘍に対する奏効率を比較し報告している。殺細胞性抗がん薬77%,sulindac+tamoxifen 50%,tamoxifen 40%,imatinib 36%,sulindac 28%であることから,腹腔内デスモイド腫瘍に対しては,殺細胞性抗がん薬が最も奏効率が高く,治療の第1選択になり得るとしている。しかしながら,どのような腹腔内デスモイド腫瘍に対し,殺細胞性抗がん薬が第1選択となり得るかいうことに関し,十分な検討は行われておらず(retrospectiveなデータや単施設での治療経験によるため),現在,標準治療は存在しない84)。
- 腹腔外デスモイド腫瘍切除後の再発率は高い(20~25%)が,術後合併症は少ない。
- 切除後の再発の原因として,不完全切除だけでなく,切除創部に新生する場合も考えられるので,腫瘍辺縁の過剰な切除は控える85)。
- 症状のない腹腔内デスモイド腫瘍に対する外科的治療は控える。(CQ4)
- 腹腔内デスモイド腫瘍による消化管通過障害には手術が考慮されるが,切除困難あるいは腸管大量切除が必要になることがある78)。
- 完全切除例とバイパスを含む非切除例との間で生存率に差はないとする報告がある86)。
- 本邦でのデータでは,腹腔外,腹腔内,混合性に対する治療は86%,48%,71%の症例で外科的切除が行われている83)。
- Churchら68)の分類を参考にして作成した腹腔内デスモイド腫瘍の病期分類を表10に示す。
表10 Churchの分類68)に準じた腹腔内デスモイド腫瘍の病期分類
| Ⅰ | Ⅱ | Ⅲ | Ⅳ | |
|---|---|---|---|---|
| 大きさ | <10 cm | 10~20 cm | 20 cm< | |
| 増大傾向 | 6カ月間増大なし | あり | あり* | |
| 尿管閉塞 | なし | あり | ||
| 腸閉塞 | なし | あり | ||
| 腫瘤触知 | なし | あり | ||
| 痛み | なし | あり | ||
| 生活制限 | なし | あり | ||
| 入院 | なし | 必要 | ||
*:6カ月以内に最大径が50%以上の増加
- 本邦での26例の解析では病期Ⅰ,Ⅲ,Ⅳが11,8,7例であり10 cm未満で症状がある病期Ⅱはまれであった。病期Ⅰでは手術をせずに非ステロイド性抗炎症剤(NSAIDs)(主にsulindac)や化学療法が多い傾向にあり,病期Ⅲになると手術が62.5%の症例に施行されており,他にもNSAIDs(主にsulindac),ホルモン治療,化学療法が高頻度で行われていた。
- 前向きな検討は行われていないが,病期Ⅰでは経過観察またはNSAIDs,病期Ⅱでは可能であれば手術およびNSAIDs+tamoxifen,病期Ⅲでは化学療法±NSAIDs±tamoxifen,病期Ⅳでは化学療法やバイパス手術などが選択肢となる。NSAIDsとしてはsulindacが主に用いられている。病期Ⅰ/Ⅱでは死亡例はなく,病期Ⅲ/Ⅳの死亡率はそれぞれ15%,44%と報告されている(図15)。尿管閉塞にはステント留置が推奨される68,87)。
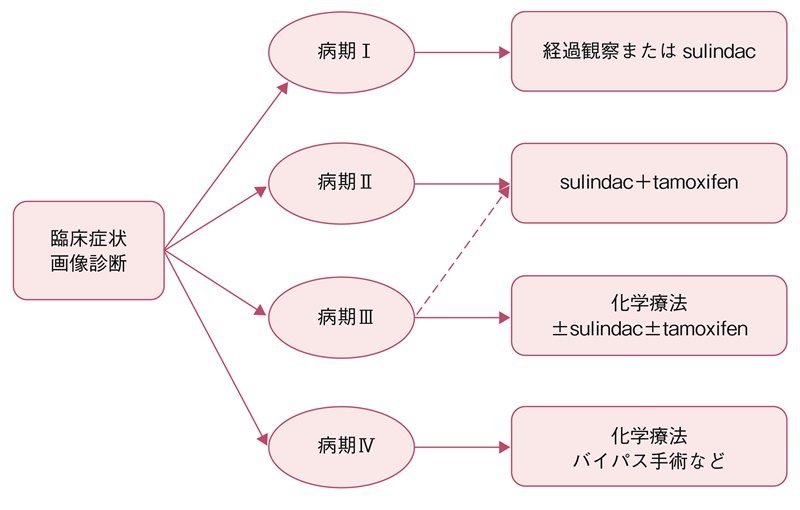
図15 腹腔内デスモイド腫瘍の病期分類と治療方針
4)その他の大腸外随伴病変
- 皮下の軟部腫瘍・骨腫などの腫瘍性病変と歯牙異常(図16)は,FAPの補助診断として参考になる。
- 非腫瘍性の先天性網膜色素上皮肥大(図17)は大腸腺腫より早期に出現する。補助診断として参考になる(サイドメモ6:先天性網膜色素上皮肥大)。
腫瘍性病変として,デスモイド腫瘍のほかに,甲状腺癌,副腎腫瘍,肝芽腫,脳腫瘍などが発生することがある(サイドメモ6:ターコット症候群)。
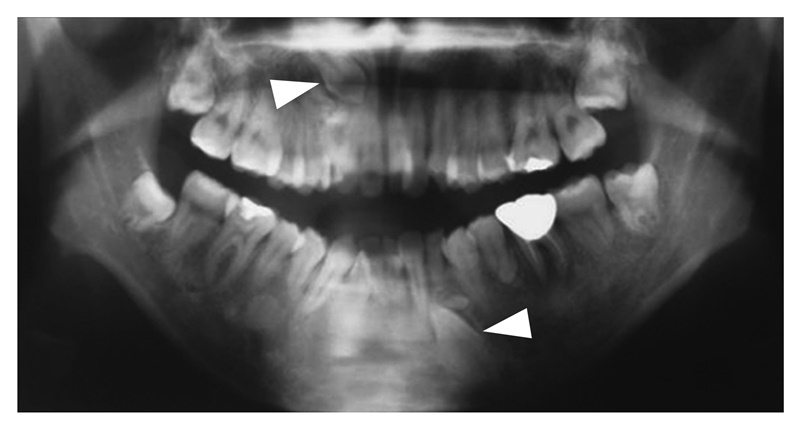
図16 歯牙異常(埋没歯▽ )
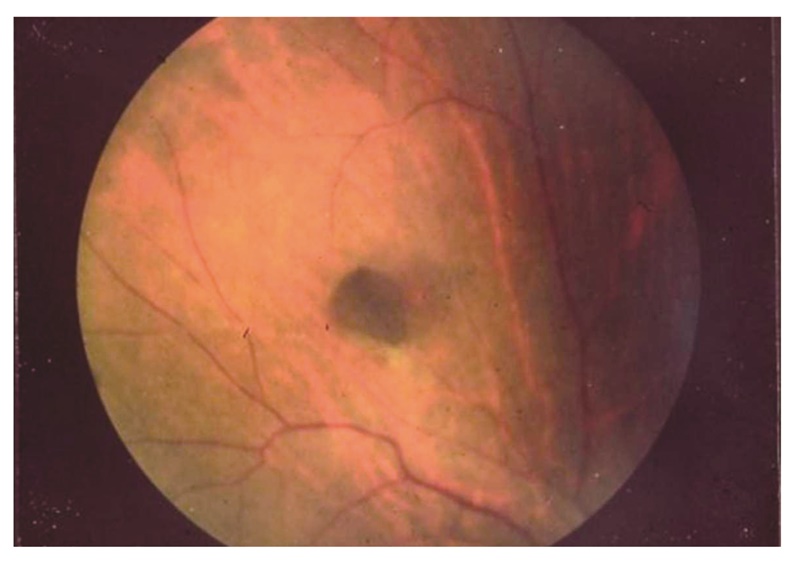
図17 先天性網膜色素上皮肥大
サイドメモ6
■先天性網膜色素上皮肥大/congenital hypertrophy of the retinal pigment epithelium:CHRPE
先天性網膜色素上皮肥大は網膜上の不連続平坦な色素性病変で,臨床症状はなく治療の必要はない。視力に影響はなく,悪性化もしない。FAP患者の約80%に合併し,生下時より認めることから,小児などのFAP補助診断に有用である。
■ターコット症候群/Turcot syndrome(type 2)
APC遺伝子バリアントを有する大腸腺腫症に脳腫瘍(主に小脳の髄芽腫)を合併するもので,ターコット症候群のtype 2に分類される(ターコット症候群type 1はリンチ症候群参照)。
4 サーベイランスと治療
1)大腸腺腫のサーベイランス
- FAPに対する下部消化管サーベイランスについて,典型的FAPでは10歳を過ぎた頃から1~2年間隔で,AFAPでは10歳代後半(18~20歳)から2~3年間隔で行うことを強く推奨する。
- 下部消化管サーベイランスを開始する年齢は,遺伝学的にFAPと診断された症例も遺伝学的検査未施行例も同様に考慮する。
- 欧州のグループが行った20歳以下のFAP患者における大腸癌発生に関する解析によれば,大腸癌の発生は10歳以前では認められず,11~15歳の間で0.2%に認められた12)。そのため,FAP患者の下部消化管サーベイランス開始の推奨年齢は10歳を過ぎた頃から考慮する25)。しかし,密生型FAPでは10歳未満でも大腸癌が発生することがある88)ため注意を要する。
- AFAPでは,典型的FAPと比較して大腸癌の発生年齢は10~15年遅く89),30歳未満での大腸癌の発生もまれであることから90),10歳代後半(18~20歳)より下部消化管のサーベイランスを開始する25)。
- 遺伝学的検査未施行例において,35歳までに大腸腺腫が認められなければFAPはほぼ否定できる。
- 一般的に,1~2年間隔(AFAPでは2~3年間隔)での下部消化管サーベイランスが推奨される。
2)大腸腺腫の治療
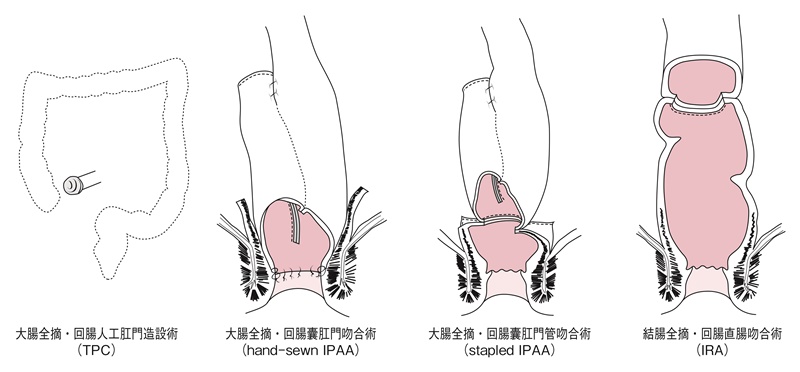
図18 FAPに対する術式
表11 FAPに対する術式の特徴
| 術式 | 大腸全摘・回腸人工肛門造設術(TPC) | 大腸全摘・回腸囊肛門(管)吻合術(IPAA) | 結腸全摘・回腸直腸吻合術 (IRA) |
|---|---|---|---|
| 利点 | 大腸癌は完全予防。 | 大腸癌はほぼ予防。 自然肛門機能の温存。 |
排便機能は良好。 比較的容易な手術手技。 IPAAよりも低い合併症率。 |
| 欠点 | 永久人工肛門による身体イメージの低下。 | 複雑な手術手技。排便機能は不安定。 吻合部近傍の肛門管部粘膜への癌発生の危険は残る。 回腸囊炎の可能性。 |
直腸癌発生の可能性(腺腫の発生状況,遺伝子バリアント部位,温存直腸の長さなどにより異なる)。 |
- 近年予防的大腸切除において腹腔鏡下手術の割合が増加している。(CQ7)
- 予防的大腸切除時に腸間膜内にデスモイド腫瘍を認める場合には,デスモイド腫瘍の再発・増大や技術的な問題からIPAAは一般的に推奨されないが,一定の条件のもとでは許容される。(各論Ⅱ 3-3):デスモイド腫瘍参照)
- 女性のFAPに対する大腸全摘術は妊孕性が低下する可能性がある。(サイドメモ8:手術と妊孕性・妊娠・出産)
- 薬物治療として非ステロイド性抗炎症剤(NSAIDs)が試みられたが,有用性は明らかでない。(CQ8)
サイドメモ7
■術式の名称
わが国では回腸囊肛門管吻合術をileoanal canal anastomosis(IACA)と呼称することが多いが,欧米では回腸囊肛門吻合術と回腸囊肛門管吻合術を区別せずに一括してileal pouch-anal anastomosis(IPAA)と呼ぶことが多い。また,回腸囊肛門吻合術をhand-sewn IPAA,回腸囊肛門管吻合術をstapled IPAAと呼ぶこともある。回腸直腸吻合術(ileorectal anastomosis:IRA)の吻合部の高さ(残存直腸の長さ)には明確な定義はなく,結腸全摘術も結腸亜全摘術も同義として扱われる。なお,大腸全摘・回腸人工肛門造設術はtotal proctocolectomy(TPC)と呼称されることが多い。
サイドメモ8
■手術と妊孕性・妊娠・出産
デンマークの女性FAP患者58名を対象とした研究95)では,妊孕性は90%で,一般集団と同等であった。162名のヨーロッパの女性FAP患者を対象とした研究では,手術を受けていないFAP患者の妊孕性は一般集団と同等であった。また,IRAを受けた患者の妊孕性も一般集団と同等であったが,IPAAを受けた患者では妊孕率が0.46倍に低下していた96)。一方,オランダのFAP患者138例を対象とした研究では,妊孕性は術式とは関連がなく,初回手術の年齢と関連があると報告されている97)。
IPAA後の妊孕性低下の原因としては,術後の癒着が考えられている。Oreslandら98)は大腸全摘後に子宮・卵管造影を行い,卵管の骨盤壁への癒着を48%に,片側閉塞を43%に,両側閉塞を10%に認めたと報告している。
FAPと潰瘍性大腸炎の患者を含めた検討では,腹腔鏡によるIPAAは,開腹手術よりも妊孕性が有意に高かったと報告されている99)。しかし,FAP患者を対象とした前向きの検討はない。
FAPと潰瘍性大腸炎の患者を含めた検討では,IPAA後の妊娠・経腟分娩は安全であることが報告されているが100,101),IPAA後の経腟分娩では会陰切開後の肛門括約筋の損傷と骨盤底筋の神経損傷を考慮する必要がある。
- 予防的大腸切除の時期については,①累積大腸癌有病率22),②腺腫密度102),③腺腫の大きさと形態,④その家系員の死亡年齢,癌発生年齢,およびデスモイド腫瘍発生状況59),⑤APC遺伝子変異部位103),⑥患者の就学,就職などの環境104),⑦回腸囊肛門(管)吻合術後の妊孕性96)や男性性機能障害105),⑧下痢,腹痛,下血などの消化管症状,および⑨腫瘍の病理組織所見,などを総合的に考慮して決定する。
- 大腸癌の有病率の点から,典型的FAPでは早ければ10歳代後期から,多くは20歳代に手術を受けることが推奨されている106,107)。
- 最近,InSiGHT(国際遺伝性消化管癌学会)は,大腸腺腫の数と大きさ(polyp burden)を用いた内視鏡的評価によるステージングを提案し(表12),下部消化管内視鏡検査の間隔や手術適応の判断がpolyp burdenと強く相関することを示した108)。また,経時的にみたpolyp burdenの明らかな増加は,臨床上の手術適応の判断基準として用いられている109)。評価者によるpolyp burdenの一致率は十分とは言えないものの,polyp burdenは下部消化管内視鏡検査の間隔や手術適応を決定し,明らかに増加する場合は,手術を考慮することが提案されている。
表12 InSiGHTポリポーシスステージングシステム
| 部位 | ステージ | ポリープ個数 | 大きさ(最大のもの) | 大きさ1 cm以上の個数 | 高度異形成または浸潤癌 | ポリペクトミー |
|---|---|---|---|---|---|---|
| 結腸 | Stage 0 | 20個未満 | 5 mm未満 | 0個 | なし | 適 |
| Stage 1 | 20~200個 | 不問 | ||||
| Stage 2 | 200~500個 | 10個未満 | ||||
| Stage 3 | 500~1000個 | 10~50個 | ||||
| Stage 4 | 1000個以上 | 不問 | あり | 不適 | ||
| 直腸 | Stage 0 | 10個未満 | 5 mm未満 | 0個 | なし | 適 |
| Stage 1 | 10~25個 | 不問 | ||||
| Stage 2 | 不問 | |||||
| Stage 3* | 25個以上 | |||||
| Stage 4* | あり | 不適 |
*鋸歯状ポリープは完全切除の可否を問わず
3)大腸癌の治療
- 進行大腸癌を伴う場合は進行大腸癌に対する標準的治療を行う。治癒が見込める場合にはFAPの病態により術式を選択する。
- 進行大腸癌を契機に発見されたFAPに対する術式は,大腸癌の進行度,部位などを考慮して総合的に決定する。治癒切除が見込める場合には領域リンパ節郭清を含む大腸全摘術や結腸全摘術も選択肢となるが,治癒切除が見込めない場合には散発性大腸癌の場合と同様の術式を選択する。
- FAPに合併する大腸癌に対する化学療法は,散発性大腸癌に対する化学療法と同様に行う。
- 大腸全摘術や結腸全摘術後においても,「大腸癌治療ガイドライン」に基づいた化学療法の選択が可能である。
- 転移巣に対しても,治癒切除が見込める場合には,散発性大腸癌に対する場合と同様に行う。
4)大腸切除前に行う大腸外随伴病変に対する検査
- 上部消化管内視鏡検査を行って,胃・十二指腸(乳頭部を含む)の腺腫や癌の有無をチェックする。
- デスモイド腫瘍の有無は触診,CTあるいはMRIでチェックする。
- 甲状腺癌(特に女性)に対する超音波検査は必ずしも大腸切除前に行う必要はないが,術後のサーベイランス計画には必ず組み込む。
- 一般的に,小腸造影や小腸内視鏡(カプセル内視鏡)検査は大腸手術前には行わない。小腸病変が疑われる症状・所見(術前画像診断を含む)がある場合には行う。(CQ10)
- 副腎腫瘍は頻度が低く,肝芽腫は2~3歳まで,脳腫瘍は青年期までに好発するので,これらの腫瘍性病変に対する術前検査は一般的に必要ない。
5)大腸切除後のサーベイランス
- 予防的大腸切除後に大腸粘膜が残存している場合には,新たに大腸癌が発生する可能性を考慮し,定期的な大腸内視鏡検査が必要である。
- 大腸癌を合併する場合,散発性大腸癌の術後と同様のサーベイランスを行う。
- 回腸直腸吻合術(IRA)後には,残存直腸の癌発生に対する長期間のサーベイランスが必要である。(サイドメモ9:結腸全摘・回腸直腸吻合術(IRA)後の直腸癌の発生リスク)
- 回腸囊肛門管吻合術(stapled IPAA)後には通常直腸粘膜が2~3 cm残存するが,回腸囊肛門吻合(hand-sewn IPAA)後でもわずかに直腸粘膜が残存する可能性がある。したがって,stapled IPAA,hand-sewn IPAAのいずれにおいても,残存直腸に対する長期間のサーベイランスが必要である。
- IPAA後の回腸囊内の腺腫発生頻度は6.7~74%と報告されている56,110-112)。また,癌が発生することも報告されているため113,114),長期間のサーベイランスが必要である。
- FAPに対するIPAA後の回腸囊炎はおよそ5%の患者に発生するが,潰瘍性大腸炎の術後よりも頻度は低い115)。臨床症状として,発熱,下痢,貧血が認められ,このような症状が出現したら,すみやかに大腸内視鏡検査を行う。
- 進行大腸癌合併例で治癒切除が行われた場合には,散発性大腸癌と同様に再発に対するサーベイランスを行う。
サイドメモ9
■結腸全摘・回腸直腸吻合術(IRA)後の直腸癌の発生リスク
IRA後の長期観察では24~43%に残存直腸に癌が発生する116,117)。IRA後20年までの経過で直腸を切除する必要があったのは,AFAPで10%,非密生型FAPで39%,密生型FAPで61%であった118)。
外科技術の進歩とともにIPAAの割合が多くなっていること91-93),直腸癌の危険因子をより多く持つ症例にIPAAが選択されることにより,IRA後の直腸切除率も40%から13%に減少し,IRA後の残存直腸癌の累積発生率も減少している119-121)。
6)大腸外随伴病変に対するサーベイランス
- 大腸切除後2~3年以内に発生しやすいデスモイド腫瘍や,十二指腸癌などの悪性腫瘍の発生を念頭においたサーベイランスが必要である。
- 治療が必要な大腸外随伴病変は大腸切除後に発生することが多い。大腸切除後の残存直腸と大腸外随伴病変に対するサーベイランスについて,表13のような方法が提唱されている13)。
表13 FAPに対する大腸切除後の残存直腸と主な随伴病変に対するサーベイランス
| 随伴病変 | 開始時期・方法 |
|---|---|
| 残存直腸腺腫 | IPAA術後は,年1回の大腸内視鏡検査と腺腫の摘除あるいは焼灼。 |
| IRA術後は,半年に1回(年齢と腺腫密度に応じる)。 | |
| 十二指腸腺腫・癌(乳頭部含む) | 大腸切除時あるいは20~25歳時のどちらか早い時期に,ベースラインの上部消化管内視鏡検査を行う。以降,腺腫の重症度に応じて定期的に繰り返す。 |
| 胃腺腫・癌 | 年1回(または十二指腸の検査と同時)の上部消化管内視鏡検査 |
| 甲状腺癌(女性) | 年1回の甲状腺の触診と超音波検査,10歳代後半から開始。 |
| 腹腔内デスモイド腫瘍 | 年1回の腹部触診。大腸切除後,特にデスモイド腫瘍の家族歴を有する場合は3年毎に腹部および骨盤のCTまたはMRI検査 |
| 脳腫瘍 | 年1回の診察。 |
| 空・回腸腺腫・癌 | 小腸の定期的な画像診断や小腸内視鏡検査は推奨されておらず,デスモイド腫瘍の画像検査(CT/MRI)の際に可及的に観察。 |
文献13)を改変
〔消化管〕- 胃底腺ポリポーシスは,通常は過形成性ポリープであり,手術の適応はない。幽門前庭部を中心に腺腫が発生する。わが国では一般集団より,FAPの方が胃癌のリスクは高い。胃のサーベイランスは十二指腸とともに行う。(CQ2)
- 十二指腸(乳頭部を含む)における癌発生頻度は高く,定期的な内視鏡による観察と腺腫に対する治療が必要である。(CQ2)
- 空・回腸に対する推奨されるサーベイランス法は確立されていない。空・回腸癌の発生はまれである。(CQ10)
- デスモイド腫瘍は大腸切除後2~3年以内に,腹壁や腸間膜,後腹膜に発生することが多い56,57)。触診,画像診断,あるいは臨床症状(腹痛,腹満,腫瘤,消化管通過障害など)に注意する。
- 悪性腫瘍としては甲状腺癌(特に女性)に注意する。年1回の触診と超音波検査を行う。(CQ9)
5 家族(血縁者)への対応
- 患者本人のほかに,家族(血縁者)にも遺伝カウンセリングを行うことが望ましい。
- 第1度近親者(親,子,兄弟姉妹)には疾患について十分な説明を行い,同意を得た上で大腸を中心とする消化管サーベイランスを行う。
- FAPを含めた遺伝性腫瘍では,家族歴の聴取が必須事項であり,家系図122,123)を用いて正確に記載・記録しておくことが望ましい(図19)。
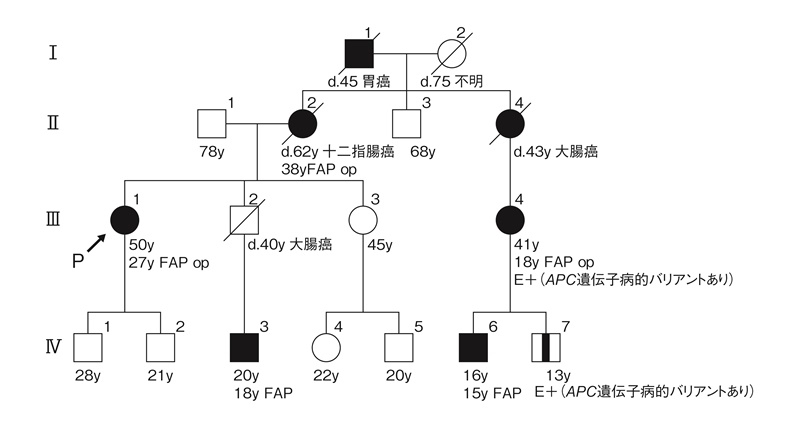
図19 FAPの家系図記載例(記載方法の要点は付録:家系図の書き方・読み方の原則参照)
家系図の記号注釈
- E+:検査で陽性(この場合APCの遺伝学的検査にて病的バリアントを検出)
 :発症前病的バリアント遺伝子保持者
:発症前病的バリアント遺伝子保持者- □13:個人の右上に個人番号を振ることがある
〔第1度近親者に対する対応〕
- 家族(血縁者)に大腸腺腫(特に複数)がある場合はFAPの診断チャート(各論Ⅰ図5:遺伝性大腸ポリポーシス 診断のフローチャート参照)に従う。
- 大腸内視鏡検査で腺腫がなければ,およそ3年毎に大腸検査を行う。
- 35歳過ぎまで複数回の大腸検査で腺腫がなければFAPはほぼ否定できる。
- 遺伝学的検査を行う場合には,検査前後に医師および,あるいは専門家による遺伝カウンセリングが必要である。
- 家系の中でAPC遺伝子の生殖細胞系列バリアントが判明していれば,血液による遺伝学的検査で診断が確定する。
- 消化管病変
- 10歳代で大腸に複数の腺腫が出現する。多くは無症状であり,時に血便,貧血,下痢,便に粘液が混じるなどの消化器症状を伴う124)。20歳未満の大腸癌の合併はきわめてまれ(FAP患者の0.2%未満)12)である。ただし消化器症状がある場合には,年齢によらず大腸内視鏡検査を行う。
- FAPと診断されている場合,大腸病変については,12~14歳以降から予防的大腸切除術が行われるまで,1~3年毎に大腸内視鏡検査を行う124--126)。
- 上部消化管病変については,小児期にも上部消化管にポリープを認めるものの,胃癌や十二指腸癌の報告はなく,原則として成人期に上部消化管内視鏡検査を開始する124-126)。
- 消化管外病変
- 肝芽腫:FAPの小児の2%未満に,主に3歳までに肝芽腫を合併する。肝芽腫のスクリーニング検査(4~6カ月毎の腹部超音波検査と血清αフェトプロテイン)を推奨する意見125,126)と,有効性のエビデンスがなく推奨できないとする意見がある124)。FAPの家族歴のある乳幼児の保護者には,肝芽腫のリスクについて説明し,保護者の希望を考慮の上,個別に対応する。
- 甲状腺癌:甲状腺癌に関し,FAPでは小児期でも発生することあるが,サーベイランスについては,成人同様確立していない。
- デスモイド腫瘍:小児期にも大腸切除術後に腹腔内デスモイド腫瘍の報告がある。
- 先天性網膜色素上皮腫大(congenital hypertrophy of the retinal pigment epithelium:CHRPE):CHRPEはFAPの患者の約80%に生下時から認める,網膜上の不連続平坦な色素性病変である。臨床症状はなく,治療の必要性はない。視力に影響なく,悪性化もしない。両側性または多発性のCHRPEは,FAP合併例に特徴的な所見である。
サイドメモ10
小児に対する遺伝学的検査と医療費助成制度
【小児に対する遺伝学的検査】
・FAPの家族歴を有する無症状の小児への,FAPの診断を目的とした遺伝学的検査については,慎重な対応が必要である。日本医学会の「医療における遺伝学的検査・診断に関するガイドライン」(2011年2月)において,未成年期に発症する疾患で発症前診断が健康管理上大きな有用性があることが予想される場合には,代諾者の了解に加えて,被検者の理解度に応じた説明を行い,本人の了解(インフォームド・アセント)を得ることが望ましいとされる。FAPの診断が確定することによる医療的ケア,心理的,社会的,経済的な益と害を被検者が理解し,意思決定に参加するように,遺伝カウンセリングが必要である。また,診断確定後にも遺伝カウンセリングを行い,継続的な支援体制を提供する。
【医療費助成制度】
・FAPは小児慢性特定疾病事業の対象疾患になっている。18歳の誕生日までに申請すると,20歳の誕生日の前日まで医療費助成が受けられる。申請のための意見書の交付にあたっては,事前に小児慢性特定疾病指定医療機関ならびに指定医の認定を要する。
Ⅲ.リンチ症候群(Lynch syndrome)
1 概 要
- リンチ症候群(Lynch syndrome)は,ミスマッチ修復遺伝子の生殖細胞系列バリアントを主な原因とする常染色体優性遺伝性疾患である(サイドメモ12:ミスマッチ修復機構)。
- 患者・家系内に大腸癌,子宮内膜癌をはじめ,さまざまな悪性腫瘍が発生する。
- 一般の大腸癌に比べ若年発症,多発性(同時性,異時性)で,右側結腸に好発し,散発性大腸癌より低分化腺癌の頻度が高い。腫瘍内リンパ球浸潤,髄様増殖,粘液癌・印環細胞癌様分化,クローン様リンパ球反応などの組織学的特徴がある6,199-201)。(各論Ⅲ 2-STEP1:MSI-H大腸癌に特徴的な病理組織学的所見参照)
- 大腸癌以外に,子宮内膜癌をはじめ,卵巣癌(CQ12),胃癌,小腸癌,胆道癌,膵癌,腎盂・尿管癌,脳腫瘍,皮膚腫瘍など多彩な悪性腫瘍(関連腫瘍)が発生する(CQ11)。近年,乳癌,膀胱癌202),前立腺癌203)についてもリンチ症候群関連腫瘍の可能性が報告されている。
- リンチ症候群における関連腫瘍の発生リスクは,原因遺伝子の種類や変異のタイプ,環境因子などにより異なる。また遺伝子バリアント保持者(以下「バリアント保持者」とする)に関連腫瘍が必ず発生するとは限らない14,199,204-209)(表15)。
表15 リンチ症候群における関連腫瘍の累積発生率(70歳まで)
| 種類 | 累積発生率 |
|---|---|
| 大腸癌 | 54~74%(男性) 30~52%(女性) |
| 子宮内膜癌 | 28~60% |
| 胃癌 | 5.8~13% |
| 卵巣癌 | 6.1~13.5% |
| 小腸癌 | 2.5~4.3% |
| 胆道癌 | 1.4~2.0% |
| 膵癌 | 0.4~3.7% |
| 腎盂・尿管癌 | 3.2~8.4% |
| 脳腫瘍 | 2.1~3.7% |
| 皮脂腺腫瘍 | 不明 |
文献14,199,204-208)より作成
〔主な原因遺伝子〕- 第3番染色体上のMLH1遺伝子
第2番染色体上のMSH2,MSH6,EPCAM各遺伝子
第7番染色体上のPMS2遺伝子
のいずれかの生殖細胞系列変異(EPCAM遺伝子の場合には3’側の欠失のみ)
- 常染色体優性遺伝
- リンチ症候群では,ミスマッチ修復遺伝子の片方のアレルに生殖細胞系列の病的バリアントを有しており,後天的にもう片方の野生型アレルに変異(あるいはプロモーター領域のメチル化)が加わるとミスマッチ修復機構が損なわれる。その結果,ゲノムの単純な反復配列であるマイクロサテライト領域に反復回数の異常(不安定性)が好発するようになる。腫瘍抑制(TGFBR2など),細胞増殖,DNA修復(MSH3,MSH6など)やアポトーシス(BAXなど)などに関わる遺伝子産物(タンパク)をコードする領域には反復配列が含まれており,これらの領域に変異が起こりやすい。
- リンチ症候群における大腸癌においても,散発性の大腸癌と同様に腺腫からがん化する経路の存在が示唆されている。詳細は不明な点も多い。(各論Ⅰ 図3:FAPとリンチ症候群の代表的ながん化のメカニズム参照)
- EPCAM(TACSTD1)遺伝子は,MSH2遺伝子の上流に隣接する遺伝子で,この遺伝子の3’側(後半部分,転写を終結するのに必要な配列)の欠失がリンチ症候群の原因となる。この欠失によりMSH2遺伝子のプロモーター領域に異常メチル化が起こり,MSH2タンパクの発現が消失する。EPCAM遺伝子欠失例では,MSH2遺伝子バリアントによるリンチ症候群と比べて,大腸癌のリスクはそれほど変わらないが,子宮内膜癌のリスクは低い210)。EPCAM遺伝子欠失は,リンチ症候群の1~3%の原因となることが報告されている211)。
- 近年の報告によると全大腸癌の0.7~3.7%8-10)占めると推定されている。
- わが国の一般集団における頻度は不明である。
サイドメモ12
■リンチ症候群の名称の変遷
1966年にHenry T. Lynchら212)が大腸癌や子宮内膜癌が多発する複数の家系を報告した。1984年にBolandら213)により癌発生が大腸癌に限られるリンチ症候群Ⅰと,大腸以外の臓器にも癌のみられるリンチ症候群Ⅱに分類され,これを区別しない場合はリンチ症候群あるいはHNPCCと呼ばれるようになった。1990年,アムステルダムで行われた国際研究グループICG-HNPCC(International Collaborative Group on HNPCC)のワークショップでHNPCCに名称が統一され,統一した基準でHNPCC家系を集積するためのアムステルダム基準(アムステルダム基準Ⅰ)214)が提唱された。1993年以降,本疾患の原因遺伝子が相次いで報告された。その結果,原因遺伝子の変異を認めてもアムステルダム基準Ⅰを満たさない家系や,アムステルダム基準Ⅰを満たしても原因遺伝子が同定されない家系が数多く認められることが判明した。そこで1998年に子宮内膜癌などの大腸癌以外の悪性腫瘍の発生を考慮した改訂アムステルダム基準(アムステルダム基準Ⅱ)(表16)がHNPCCの共同研究目的に提唱された215)。その後,HNPCCの名称について繰り返し検討された結果,大腸以外の臓器にさまざまな悪性腫瘍が発生する本疾患の特徴を踏まえ,HNPCCの名称ではふさわしくないと考えられるようになった。現在は報告者のLynch博士の名にちなんでリンチ症候群の名称を用いることが多くなっている。
■ミスマッチ修復機構
細胞はDNA複製の際に生じた誤った塩基対合(ミスマッチ)を発見し,修復する働きをもつ。ミスマッチ修復機構が損なわれるとミスペアや単純繰り返し配列の挿入・欠失の頻度が10~1,000倍高くなり,マイクロサテライト領域の不安定性を生じる(サイドメモ14:MSI検査の方法と結果の評価)
■Germline epimutaion
近年,リンチ症候群の一部で,腫瘍発生にエピミューテーション(epimutation)が関与していることが明らかにされた。エピミューテーションとは,塩基配列には変化がないが,DNAのメチル化異常など遺伝子発現に関わる分子の修飾により遺伝子発現に変化をもたらす現象である。まれではあるが,生殖細胞系列のMLH1遺伝子のプロモーター領域の異常メチル化がリンチ症候群の原因になることが報告されている216)。
2 診 断
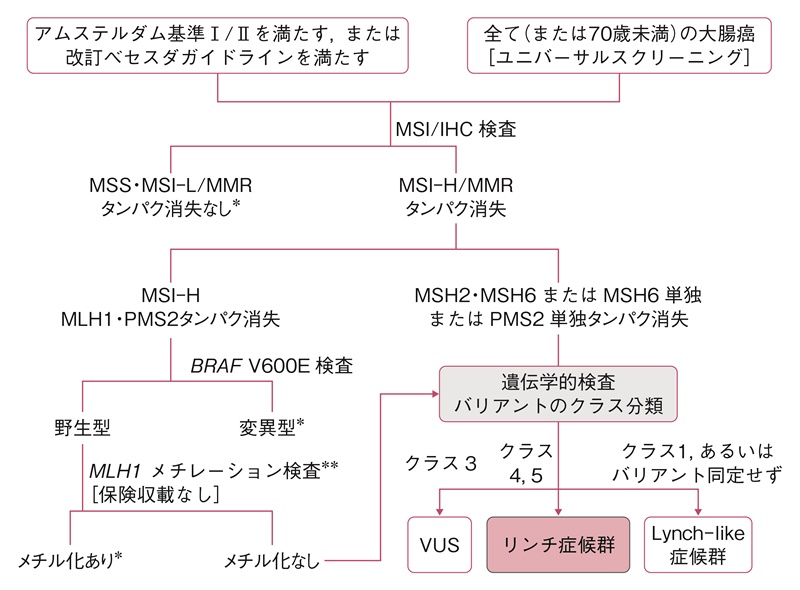
図21 リンチ症候群の診断手順
MSI:microsatellite instability(マイクロサテライト不安定性),IHC:immunohistochemistry(免疫組織化学的染色),MSI-H:high-frequency MSI(高頻度MSI),MSI-L:low-frequency MSI(低頻度MSI),MSS:microsatellite stabe(マイクロサテライト安定性),MMR:mismatch repair(ミスマッチ修復),VUS:variant of uncertain significance(病的か不明なバリアント)。
*:遺伝学的検査に進まない,**:BRAF V600E検査を行わずにMLH1メチレーション検査のみを行っても良い。
1)診断の流れ
- リンチ症候群が疑われる臨床病理学的情報(家族歴を含む)を有する患者に対し,以下のSTEP 1からSTEP 3の手順で確定診断する(図21)。
- STEP 1:アムステルダム基準Ⅱ215)(表16,図22A,22B)あるいは改訂ベセスダガイドライン217)(表17)を満たすかを確認する(第1次スクリーニング)。ユニバーサルスクリーニングでは,全て(あるいは70歳以下)の大腸癌や子宮内膜癌がSTEP 2に進む。(CQ14)
- STEP 2:腫瘍組織のマイクロサテライト不安定性(microsatellite instability:MSI)検査,あるいは原因遺伝子産物に対する免疫組織学的検査を行い,高頻度MSI(high-frequency MSI:MSI-H)または免疫染色でミスマッチ修復タンパクの消失を確認する(第2次スクリーニング)。(サイドメモ13:リンチ症候群のスクリーニング検査におけるMSI検査の注意点,サイドメモ14:MSI検査の方法と結果の評価)
MSI-HまたはMLH1,PMS2の発現消失を示す症例で,腫瘍組織がBRAF V600Eバリアント,あるいはMLH1プロモーターメチル化陽性であれば,STEP 3に進まなくてよい。 - STEP 3:確定診断として,ミスマッチ修復遺伝子の生殖細胞系列における病的変異を同定する(保険収載されていない)。(CQ15)
- ユニバーサルスクリーニング:
近年,欧米では全て(あるいは70歳以下)の大腸癌や子宮内膜癌に対し,MSI検査やミスマッチ修復タンパクに対する免疫染色を行うユニバーサルスクリーニングがリンチ症候群の診断に関し,感度と費用対効果の高い方法として推奨されている。 - MSI検査,ミスマッチ修復タンパク質に対する免疫染色,および両者の併用によるスクリーニング感度は,近年のプール解析においてそれぞれ0.93(95%信頼区間:0.87~0.96),0.91(95%信頼区間:0.85~0.95),0.97(95%信頼区間:0.90~0.99)と,いずれも高い感度が示されている218)。
- ユニバーサルスクリーニングから得られた海外からの報告によるリンチ症候群の頻度は2.4~3.7%と報告されている8,9)。
- 高齢の大腸癌患者では,リンチ症候群患者が含まれる割合が相対的に低い一方,散発性ミスマッチ修復異常大腸癌の頻度が高い傾向がある10,219,220)。このため,スクリーニングの効率と費用対効果を考慮し,大腸癌患者全例ではなく,70歳未満など,一定の年齢以下の患者を対象としてスクリーニングを行うことも提唱されている。
- 欧米ではリンチ症候群のスクリーニング検査としてのMSI検査,免疫染色の施行には患者の個別同意が必要ないと考えられているが,国内ではリンチ症候群のスクリーニング検査を行うことに関して事前説明を行うことが望ましいとされている221)。
STEP 1 第1次スクリーニングに用いる基準
表16 アムステルダム基準Ⅱ(1999)215)
- 1人の罹患者はその他の2人に対して第1度近親者である。
- 少なくとも連続する2世代で罹患している。
- 少なくとも1人の癌は50歳未満で診断されている。
- 腫瘍は病理学的に癌であることが確認されている。
- FAPが除外されている。
表17 改訂ベセスダガイドライン(2004)217)
- 50歳未満で診断された大腸癌。
- 年齢に関わりなく,同時性あるいは異時性大腸癌あるいはその他のリンチ症候群関連腫瘍*がある。
- 60歳未満で診断されたMSI-Hの組織学的所見**を有する大腸癌。
- 第1度近親者が1人以上リンチ症候群関連腫瘍に罹患しており,そのうち一つは50歳未満で診断された大腸癌。
- 年齢に関わりなく,第1度あるいは第2度近親者の2人以上がリンチ症候群関連腫瘍と診断されている患者の大腸癌。
*:大腸癌,子宮内膜癌,胃癌,卵巣癌,膵癌,胆道癌,小腸癌,腎盂・尿管癌,脳腫瘍(通常はターコット症候群にみられるglioblastoma),ムア・トレ症候群の皮脂腺腫や角化棘細胞腫
**:腫瘍内リンパ球浸潤,クローン様リンパ球反応,粘液癌・印環細胞癌様分化,髄様増殖
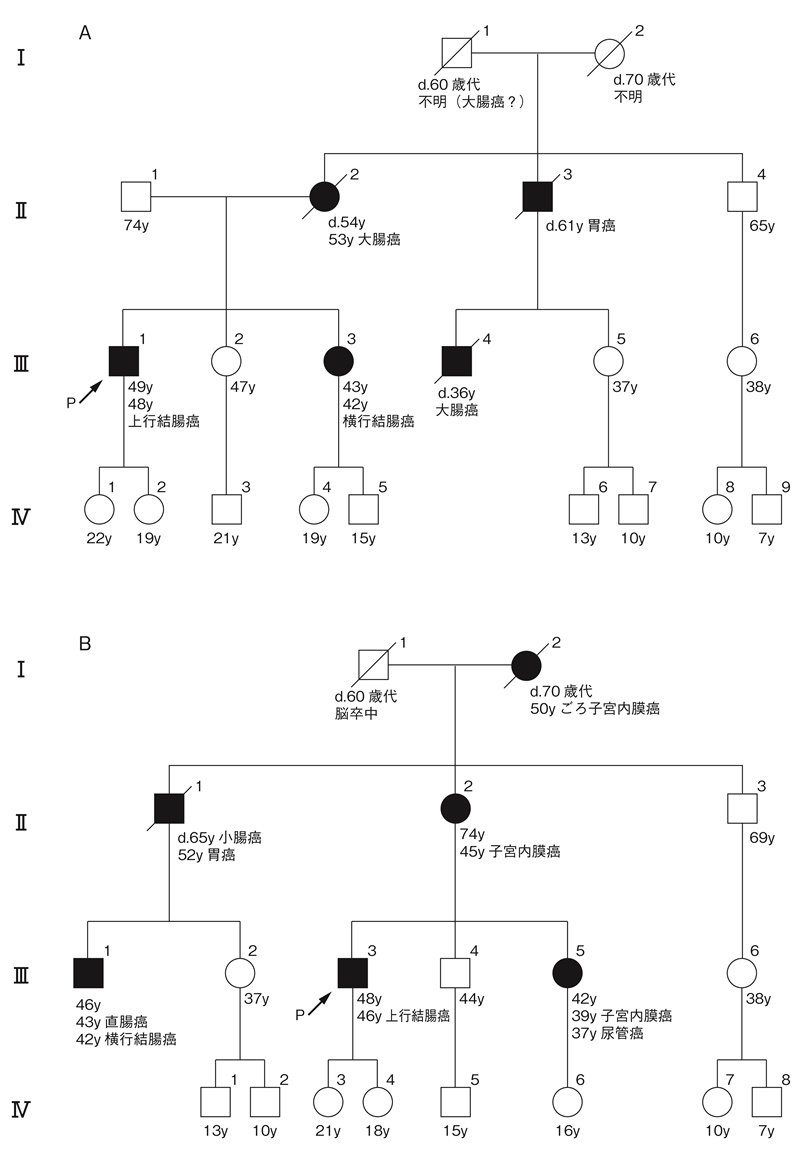
図22 アムステルダム基準Ⅱ215)に合致する家族歴(付録:家系図の書き方・読み方の原則参照)
A:大腸癌の多発家系
B:大腸癌以外の関連腫瘍多発家系
- リンチ症候群の家系のなかで,アムステルダム基準Ⅱ215)を満たす家系は27~41%212,219),改訂ベセスダガイドライン217)を満たす家系は68~89%と報告されており,改訂ベセスダガイドラインの方がより多くのリンチ症候群を拾い上げられる219)。
- 大腸癌患者の約1/4が改訂ベセスダガイドラインを満たす222)。すなわち,リンチ症候群ではない散発性大腸癌でも改訂ベセスダガイドライン217)を満たすものが少なくない。
- 大腸癌研究会のプロジェクト研究では,全大腸癌患者の1.2%がアムステルダム基準Ⅱを満たした223)。
MSI-H大腸癌に特徴的な病理組織学的所見:
MSI-H大腸癌は非MSI-H大腸癌と比べ,いくつかの組織学的特徴が有意に多く認められるため,これらの所見がリンチ症候群疑い患者の拾い上げに有用である。改訂ベセスダガイドライン217)においては,①腫瘍内リンパ球浸潤(tumor infiltrating lymphocytes:TIL),②髄様増殖,③粘液癌・印環細胞癌様分化,④クローン様リンパ球反応(Crohn’s-like lymphocytic reaction)の4項目が挙げられている(図23A,23B,23C,23D)。ただし,これらの組織学的特徴は必ずしもリンチ症候群に特有のものではなく,散発性MSI-H大腸癌にも共通して認められる224)。
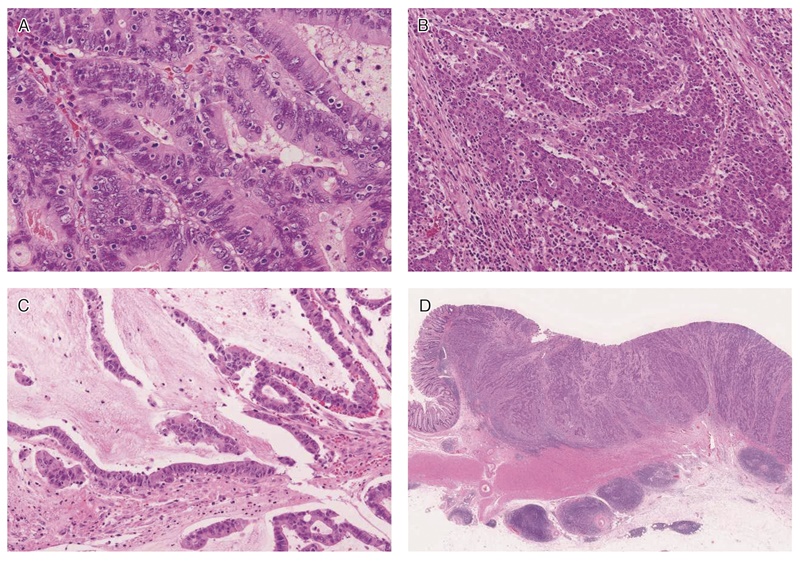
図23 MSI-H大腸癌の組織学的特徴
A:腫瘍内リンパ球浸潤。腫瘍上皮内にhaloを伴ったリンパ球浸潤を認める。
B:髄様増殖。腫瘍細胞は腺管を形成せず,充実性胞巣状の増殖を示す。
C:粘液癌・印環細胞癌様分化。多量の細胞外粘液を伴う。
D:クローン様リンパ球反応。腫瘍周囲に多数のリンパ球の集簇巣を認める。
STEP 2 第2次スクリーニングで行う検査
MSI検査:
ミスマッチ修復機構に異常がある腫瘍細胞では,ゲノムの中に存在する1~数塩基の繰り返し配列であるマイクロサテライトが正常細胞とは異なる反復回数を示すことがある。この現象をマイクロサイト不安定性(MSI)という。
リンチ症候群の大腸癌の90%以上に高頻度マイクロサテライト不安定性(MSI-H)を認めることが報告されている225)。一方,大腸癌全体に対するMSI-Hの割合は,欧米の報告では12~16%225-227),わが国の報告では6~7%である228,229)。そのため,MSI検査はリンチ症候群を疑う症例を絞り込むスクリーニング検査として有用である。臨床情報からリンチ症候群が疑われ,腫瘍(検出感度は落ちるが大腸腺腫でも可)のMSI検査の結果がMSI-Hであれば,リンチ症候群が強く疑われる。
MSI検査は,リンチ症候群が疑われる大腸癌症例を対象とする悪性腫瘍遺伝子検査として平成18年より保険収載された。検査の実施に際しては遺伝性のがんである可能性について,十分な説明と同意が必要である。日本遺伝性腫瘍学会ホームページ(http://jsht.umin.jp/project/data/index.html)のリンク先に参考資料が公開されている(サイドメモ14:MSI検査の方法と結果の評価)。
サイドメモ13
■リンチ症候群のスクリーニング検査におけるMSI検査の注意点
MSI検査で注意すべき点として,MSH6遺伝子に生殖細胞系列変異があるリンチ症候群では,MSI-Hを示さないことがある230,231)。したがって,MSI-L,MSSであってもアムステルダム基準Ⅱ215)を満たしている場合やリンチ症候群を強く疑う臨床的特徴(若年発症や多重がんなど)が認められる場合は,ミスマッチ修復遺伝子の遺伝学的検査を考慮する13)。
サイドメモ14
■MSI検査の方法と結果の評価
MSI検査には腫瘍部の凍結材料またはホルマリン固定パラフィン包埋標本が必要である。抽出したDNAから,一般に5種類の1塩基繰り返しマーカー(プロメガパネル:BAT-25,BAT-26,NR-21,NR-24,MONO-27)を用いて,腫瘍組織のマイクロサテライト不安定性を判定する(図24)。マイクロサテライトの長さが変化している場合をMSIと判定し,2つ以上のマーカーがMSIを示す場合をMSI-H(high-frequency MSI),1つのマーカーがMSIを示す場合をMSI-L(low-frequency MSI),いずれのマーカーもMSIを示さない場合をMSS(microsatellite stable)とする(図24)。
なお,従来用いられていたベセスダマーカー(1塩基の繰り返しマーカー2種類と,2塩基の繰り返しマーカー3種類からなる)などではマイクロサテライトの長さを判定する対照として正常組織を必要としたが,プロメガパネルに用いられる1塩基繰り返しマーカーでは個体間の差異が少ないため(quasi-monomorphic mononucleotide),腫瘍組織のみで判定可能である。
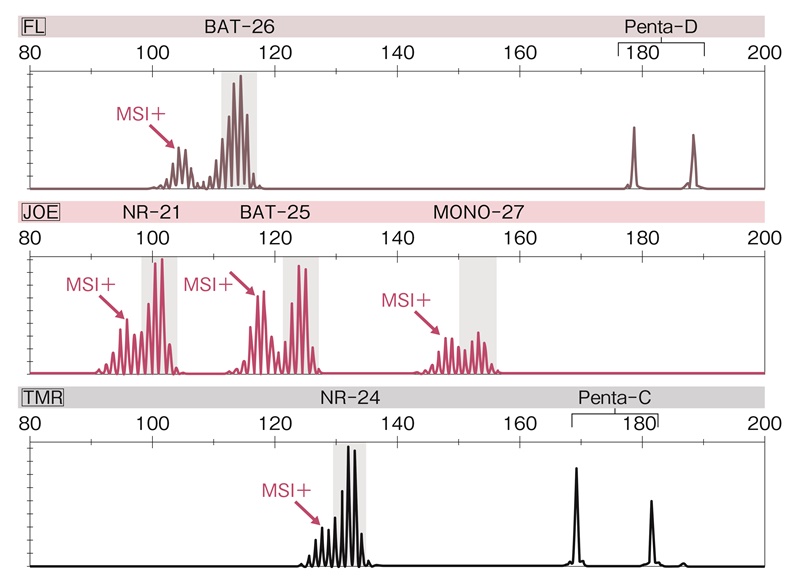
図24 プロメガパネルを用いたMSIの解析例
5種類の1塩基繰り返しマーカーの全て(BAT-26,NR-21,BAT-25,MONO-27,NR-24)で,腫瘍組織のマイクロサテライト長が基準と異なり,MSI-Hと判定される。
免疫組織化学的染色(免疫染色,immunohistochemistry:IHC):
リンチ症候群関連腫瘍の大半で,ミスマッチ修復遺伝子であるMLH1,MSH2,MSH6,PMS2のいずれかの遺伝子の両アレルに不活化が起きており,その大部分の症例で対応するタンパクの発現が消失する。MSI-Hはミスマッチ修復機能の異常を原因とするため,MSI検査とミスマッチ修復タンパクに対する免疫染色の結果は高い一致率を示す。大腸癌におけるMSI検査と免疫染色の一致率は90%,リンチ症候群のスクリーニングにおける免疫染色の偽陰性率は5~10%と報告されている160,185)。MSI検査に対する免疫染色の利点は,多くの施設で実施可能であることと,原因遺伝子を推定できることである(サイドメモ15:例外的な染色結果)。
免疫染色の実施に際してはMSI検査同様十分な説明と同意が必要である。日本遺伝性腫瘍学会ホームページ(http://jsht.umin.jp/project/data/index.html)のリンク先に参考資料が公開されている。
MSI検査と免疫染色は感度・特異度は同等であるが,各々の検査のコストや利便性は施設ごとに異なるので,施設の検査体制も加味して総合的に判断し,どちらか一方の検査を選択すればよい。なお,一方の検査が陰性であっても臨床的にリンチ症候群が疑わしい場合には,もう一方の検査を行うことで相互補完的なスクリーニングが可能となる。
●染色評価にあたっては内部陽性対照を用いて染色の適切性を確認しておく。
1.内部陽性対照
ミスマッチ修復タンパクは核に局在し,増殖細胞により強く発現する。非腫瘍組織では大腸粘膜の腺底部やリンパ濾胞の胚中心がよい陽性コントロールになる(図25)。腫瘍組織は一般に増殖活性が高いため,内部陽性コントロールの染色が確認できれば判定は容易なことが多い。
2.染色のパターンと評価
ミスマッチ修復異常のない腫瘍では4種類のタンパク全てが発現している。ミスマッチ修復異常を呈する腫瘍では異常のあるミスマッチ修復遺伝子を反映したタンパクの発現消失を呈するが,個々のミスマッチ修復遺伝子異常とタンパクの発現消失は1対1対応にならない(表18,図26)。大半の症例は表18に当てはまる染色パターンを示す。表18に当てはまらない染色結果を得た場合は,例外的な症例である可能性を考慮する前に染色の妥当性を確認すべきである。浸潤がんの場合,原則として発現消失はびまん性である。
MLH1遺伝子バリアントを有する腫瘍はMLH1に加えてPMS2,MSH2遺伝子バリアントを有する腫瘍はMSH2に加えてMSH6の発現消失を伴うため(表18),PMS2,MSH6に対する2種類の抗体のみで4抗体を用いた場合と同等の感度でリンチ症候群のスクリーニングを行うことが可能である232)。PMS2の発現消失が認められた場合はMLH1の染色を,MSH6の発現消失が認められた場合はMSH2の染色をそれぞれ追加し,原因となっている遺伝子バリアントの種類の推定を行う。
表18 ミスマッチ修復タンパクに対する免疫染色パターンと推定される原因遺伝子の種類
| 免疫染色での発現 | |||||
|---|---|---|---|---|---|
| MLH1 | MSH2 | PMS2 | MSH6 | ||
| 推定される原因遺伝子 | MLH1 | - | + | - | + |
| MSH2 | + | - | + | - | |
| PMS2 | + | + | - | + | |
| MSH6 | + | + | + | - | |
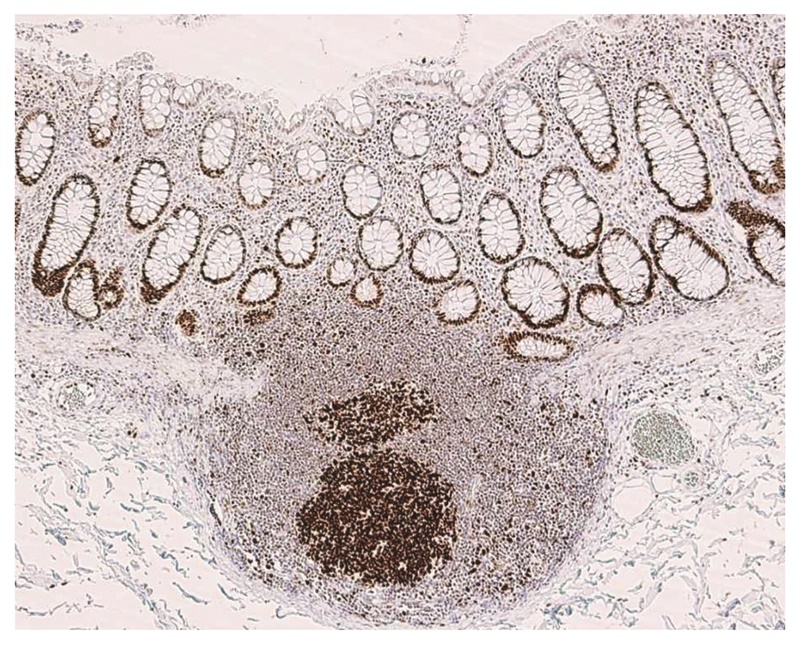
図25 非腫瘍粘膜におけるMSH2染色例
リンパ濾胞胚中心と非腫瘍陰窩の腺底部に強い染色を認める。
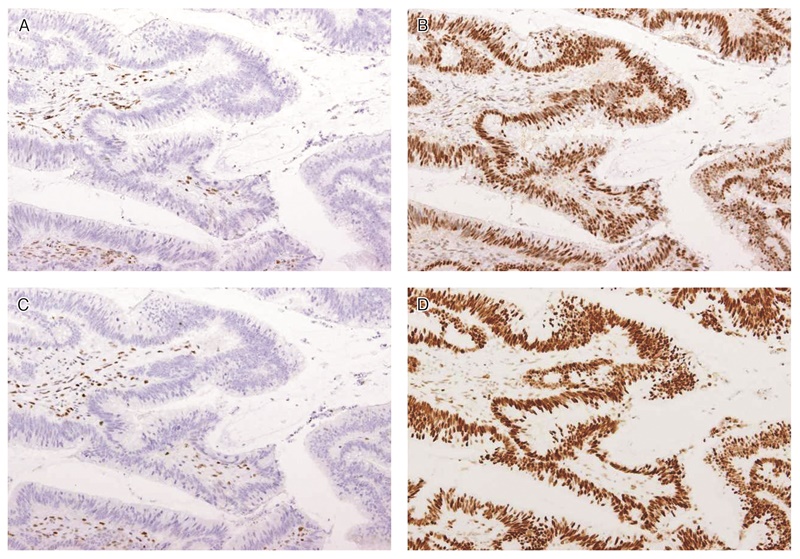
図26 MLH1遺伝子変異リンチ症候群に伴う大腸癌におけるミスマッチ修復タンパク染色例
MLH1(A),PMS2(C)の発現消失を認める。MSH2(B),MSH6(D)の発現は保たれている。いずれの染色においても内部陰性対照となる間質細胞の陽性所見が認められる。
サイドメモ15
例外的な染色結果
■ミスセンスバリアントなどによる異常タンパクの発現
ミスセンスバリアントの場合,機能が保たれていないタンパクが発現することがある。MLH1バリアントを伴うリンチ症候群に比較的多いことが知られており,これらの症例の大半はPMS2の単独発現消失を呈する232)。ただし,免疫染色で異常が全く指摘できない例がまれに存在する。免疫染色で異常が認められなくても臨床的にリンチ症候群が強く疑われる場合は,MSI検査を追加することで拾い上げが可能となることがある。
■マイクロサテライト不安定性によるミスマッチ修復遺伝子の2次的バリアント
ミスマッチ修復遺伝子のいくつかには繰り返し配列を持つものがあり,2次的なバリアントを起こすことがある。MLH1バリアント(MLH1/PMS発現消失)例では,びまん性または領域性にMSH6の発現消失をきたすことがある233)。
■術前化学放射線療法によるMSH6の発現消失
術前化学放射線療法を行った場合,MSH6に異常がなくてもMSH6の発現消失を示すことが報告されている233)。
BRAF V600E検査・MLH1プロモーターメチル化検査:
ミスマッチ修復異常を示す散発性大腸癌の大半はMLH1プロモーターメチル化によるMLH1発現抑制を原因としている。MSI-HまたはMLH1,PMS2の発現消失を示す症例では,腫瘍組織のBRAF V600Eバリアント,MLH1プロモーターメチル化の検索を行う234)。BRAF V600Eバリアント,あるいはメチル化が陽性であればリンチ症候群はほぼ否定できるため,STEP 3に進まなくてよい患者を選択することができる。BRAF V600E遺伝子バリアントの検査は平成30年より保険収載されている。
- MLH1プロモーターメチル化検査はBRAF V600Eバリアント検査よりもより高い感度で散発例を除外することが可能であるため,海外のガイドラインの多くではメチル化検査が推奨されている13,126,235-238)。
- MLH1プロモーターメチル化検査は保険収載されておらず,国内では受託検査としても提供されていないため,この検査による生殖系列細胞バリアント検査の前段階での散発性MSI-H大腸癌症例除外を行うことが難しい。
- PMS2遺伝子にバリアントがあるリンチ症候群の大腸癌の一部ではBRAF V600E遺伝子バリアントを認めることが報告されており239),注意が必要である。
STEP 3 確定診断のための検査
- ミスマッチ修復遺伝子などの遺伝学的検査:
患者の血液を用いて,ミスマッチ修復遺伝子とEPCAM遺伝子の生殖細胞系列におけるバリアント(EPCAM遺伝子では3’側の欠失のみ)の有無を直接検査する。病的バリアントが同定されれば,リンチ症候群の確定診断とする。わが国では保険収載されておらず,全額自己負担もしくは研究として実施しているのが現状である(遺伝学的検査は検査会社に依頼可能)。本検査の前後には必ず遺伝カウンセリングを行う。(各論Ⅰ 2-3):遺伝カウンセリング参照) - スクリーニングの過程でミスマッチ修復遺伝子の遺伝学的検査に至らなかった場合や,遺伝学的検査を実施したが原因遺伝子の病的バリアントが検出されなかった場合でも,リンチ症候群の可能性が残る。
- 臨床的にリンチ症候群の特徴が強く出ている家系に対しては,MSI検査や免疫染色によるスクリーニングを経ずに,直接ミスマッチ修復遺伝子の遺伝学的検査を行うこともある。
- ミスマッチ修復遺伝子の遺伝学的検査は,家系のなかでもリンチ症候群の臨床的特徴(大腸癌や子宮内膜癌などの多重がん,若年発症など)を持った個人に実施することが望ましい。
サイドメモ16
■ムア・トレ症候群/Muir-Torre syndrome
大腸癌をはじめとする種々のリンチ症候群関連腫瘍に皮脂腺腫瘍(皮脂腺腫,皮脂腺上皮腫,皮脂腺癌)や角化棘細胞腫などを合併する疾患。主にMSH2遺伝子の生殖細胞系列バリアントが認められる240)。
■ターコット症候群/Turcot syndrome(type 1)
リンチ症候群の関連腫瘍として大腸癌と脳腫瘍,主に神経膠芽腫を合併する疾患。MLH1,PMS2遺伝子の生殖細胞系列バリアントやプロモーター領域のメチル化が認められる241)。脳腫瘍はリンチ症候群の主要な死因と報告されており242),注意が必要である(CQ9:ターコット症候群type 2)。
■Constitutional mismatch repair deficiency:CMMRD
CMMRD症候群36)は,生殖細胞系列でミスマッチ修復遺伝子の両アレルに病的バリアントを有し,常染色体劣性遺伝形式をとる。リンチ症候群とは,がん発生スペクトラムや好発年齢が異なる。CMMRD症候群の91家系146症例のうち85症例(58%)は,PMS2の双方の対立遺伝子バリアントが原因であった37)。146症例中,悪性腫瘍は,81例(55%)で中枢神経腫瘍(中央値9歳),59例(40%)で大腸癌(中央値16歳),48例(33%)で血液腫瘍(中央値6歳)であった。さらに特徴的なことに80%以上で神経線維腫症1型(neurofibromatosis type 1:NF1)類似のカフェ・オ・レ斑(Café au lait spots)が認められたという報告37)がある。
2)鑑別を要する疾患
散発性MSI-H大腸癌:
MSI-Hを示す散発性大腸癌は,高齢女性,低分化腺癌,右側結腸に多い,などの特徴を認める。MSI-Hを示す主な原因はMLH1遺伝子のプロモーター領域の後天的な異常メチル化である243)。このような腫瘍では免疫染色でMLH1タンパクの発現消失を認める。またMSI-Hを示す大腸癌の35~43%に腫瘍組織のBRAF V600E遺伝子バリアントを認めるが244,245),リンチ症候群の大部分の大腸癌はMSI-Hを示しても,BRAF V600E遺伝子はほとんど検出されない246)。したがって,BRAF V600E遺伝子バリアントの有無が両者の鑑別に利用されることがある。
ポリメラーゼ校正関連ポリポーシス(polymerase proofreading-associated polyposis:PPAP):
PPAP37-39)は,FAP(AFAP)とリンチ症候群に類似した病態を示すことがあり,鑑別を要する(各論Ⅱ 2-3):鑑別を要する疾患参照)。POLEを原因遺伝子とするPPAPの大腸癌ではMSI-Hを示すことがある。
家族性大腸癌タイプX:
アムステルダム基準Ⅰ214)注2を満たすが,ミスマッチ修復遺伝子の生殖細胞系列バリアントが認められない,あるいは大腸癌がMSI-Hでない場合,リンチ症候群ではない可能性が高く,家族性大腸癌タイプX247)の名称が提唱されている。複数の疾患群からなると推測されている。大腸癌以外のリンチ症候群関連腫瘍のリスクは有意に低いことが,欧米や日本248)から報告されている。
注2 アムステルダム基準Ⅰ:アムステルダム基準Ⅱは大腸癌,子宮内膜癌,腎盂・尿管癌,小腸癌を関連腫瘍とするが,アムステルダム基準Ⅰ214)では大腸癌のみを関連腫瘍とする。
Lynch-like syndrome:
ミスマッチ修復異常(MSI-Hまたはミスマッチ修復タンパクの発現消失)を示す大腸癌の中でMLH1プロモーターメチル化を認めず,かつリンチ症候群の原因となるミスマッチ修復遺伝子やEPCAMの病的バリアントも認めない症例は,Lynch-like syndromeと呼ばれている。その原因として,①ミスマッチ修復遺伝子の両アレル生じた体細胞バリアント,②同定できない生殖細胞系列のミスマッチ修復遺伝子バリアント,③MMR遺伝子以外の生殖細胞系列バリアント,などが挙げられている。未だ不明な点も多い249,250)。
3 治 療
1)大腸癌の治療
- リンチ症候群の大腸癌に対する大腸の切除範囲(術式)として,以下の選択肢がある。
(1)散発性大腸癌と同等の切除範囲
(2)結腸全摘術
(3)大腸全摘術 - 予防的大腸切除の有用性についてコンセンサスはなく,一般的には推奨されない。
- リンチ症候群の大腸癌は,同時性・異時性を問わず,多発する傾向があるので,手術の前には全大腸を検査する。
- 欧米ではリンチ症候群の大腸癌に対し,結腸癌に対する結腸全摘術,直腸癌に対する大腸全摘術などの拡大手術を推奨する報告がある。(CQ16)
- リンチ症候群の未発症バリアント保持者に対する予防的大腸切除術の有効性は検討されておらず,一般的に推奨されない。(CQ16)
- リンチ症候群の大腸癌の大部分がMSI-Hの特徴を示す。MSI-Hの大腸癌は一般的に5-fluorouracil(FU)系抗がん薬の効果が認められないことが報告されているが,リンチ症候群の大腸癌に限定した化学療法の有用性については明らかになっていない。(CQ17,CQ18)
2)大腸癌以外の関連腫瘍への対応
(1)消化器腫瘍(胃癌,小腸癌,胆道癌,膵癌など)
(2)婦人科腫瘍(子宮内膜癌,卵巣癌など)(CQ12,CQ13)
(3)泌尿器腫瘍(腎盂・尿管癌など)
(4)その他(脳腫瘍,皮膚腫瘍など)
- (1)~(4)のうち,婦人科癌を除けば,リンチ症候群に対する治療上の特別な配慮については明らかなエビデンスはなく,通常の散発性癌(腫瘍)と同様の治療が行われているのが現状である。
- 大腸癌を合併したリンチ症候群に対しては,大腸手術の術前に関連腫瘍(特に婦人科癌,泌尿器癌,大腸癌以外の消化器癌)のスクリーニングを行っておくことが望ましい。
4 術後のサーベイランス
1)大腸多発癌のサーベイランスと腺腫の摘除
- リンチ症候群の大腸癌の術後には,異時性多発癌の発生に留意し,生涯にわたって定期的な大腸内視鏡検査が必要である。(CQ12)
- 切除した大腸癌の再発に関するサーベイランスは,散発性大腸癌に準ずる(「大腸癌治療ガイドライン」参照)。
- 大腸腺腫は大腸癌の原因になるので,発見した場合は摘除する。
2)大腸癌以外の関連腫瘍のサーベイランス
- リンチ症候群の主な関連腫瘍に対するサーベイランスについては表19のような方法が,欧州の専門家グループにより提唱されている235)。
表19 リンチ症候群の主な関連腫瘍に対するサーベイランスの目安
| 部位 | 検査方法 | 検査開始年齢 | 検査間隔 | コメント | 文献 |
|---|---|---|---|---|---|
| 大腸 | 大腸内視鏡 | 20~25歳 | 1~2年 | 13,126,235-238) | |
| 子宮・卵巣 | 経腟US,子宮内膜組織診(または細胞診),(CA-125) | 30~35歳 | 1年 | 13,126,235-238) | |
| 胃・十二指腸 | HP感染 | 30~35歳 | HP感染があれば除菌 | 13,126,235-237) | |
| 上部消化管内視鏡 | 30~35歳 | 1~3年 | 胃癌リスクの高い集団,または胃・十二指腸癌の家族歴がある場合に考慮 | 13,126,235-237) | |
| 尿路 | 検尿(または尿細胞診) | 30~35歳 | 1年 | MSH2バリアント,または尿路上皮癌の家族歴がある場合に考慮 | 13,235-238) |
US:超音波断層法(ultrasonography)
HP:ヘリコバクター・ピロリ(Helicobacter pylori)
- ヘリコバクター・ピロリ(Helicobacter pylori:HP)感染胃炎のスクリーニングと感染者に対する除菌が提唱されている。東アジアのように胃癌の多い地域や,胃癌の家族歴を有するリンチ症候群の患者と血縁者には,上部消化管内視鏡検査によるサーベイランスを1~3年毎に行うことが提唱されている251)。
- 子宮内膜癌と卵巣癌の定期的なサーベイランス法やその施行間隔についてはコンセンサスが得られていない。(CQ12)
- 泌尿器系の関連腫瘍としては腎盂・尿管癌が挙げられる。MSH2遺伝子の生殖細胞系列にバリアントを有する患者に多いとされているが,定期的な検尿,尿細胞診を含め,予後の改善に有用性が証明されたサーベイランス法はない。
5 リンチ症候群であることが確定していない大腸癌患者への対応
1)遺伝学的検査を実施していない場合
- リンチ症候群が疑われても,遺伝学的検査による確定診断がなされていない患者には,臨床情報,MSI検査やMMRタンパクのIHC検査の結果などからリンチ症候群の可能性を個別に評価し,関連腫瘍のサーベイランスを行う(図27)。
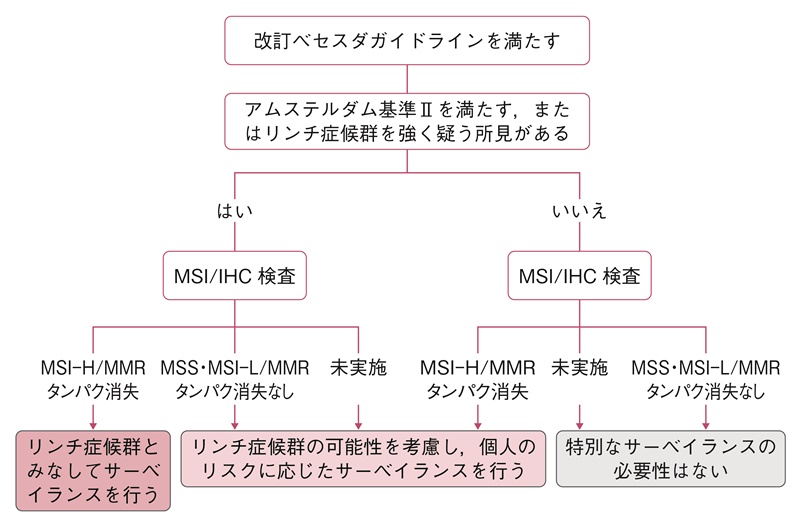
図27 リンチ症候群であることが確定していない患者への対応
2)遺伝学的検査の結果がVUSであった場合
- リンチ症候群の遺伝学的検査を行った結果がVUSであった患者には(図21),家系内の発がん状況に応じたサーベイランスを提案する。濃厚な家族歴がない場合には,特別なサーベイランスの必要はなく,散発性大腸癌と同様に一般検診を行う。
3)リンチ症候群を強く疑う家族歴があるが遺伝学的検査で確定しない場合
- リンチ症候群を疑う濃厚な家族歴があるにもかかわらず遺伝学的検査で病的バリアントが確認されない大腸癌患者には,リンチ症候群とみなしてサーベイランスを行う。
- 「アムステルダム基準Ⅱを満たす」,または「リンチ症候群を強く疑う既往歴・家族歴がある」場合で,MSI検査の結果がMSI-H,またはIHC検査の結果がMMRタンパク消失であれば,遺伝学的検査が未施行でもリンチ症候群としてサーベイランスを行う。
- 「アムステルダム基準Ⅱを満たす」,または「リンチ症候群を強く疑う既往歴・家族歴がある」場合で,MSS/MSI-LまたはMMRタンパク消失なし(ミスマッチ修復遺伝子異常を強く疑わせる所見がない)の場合でも,リンチ症候群が否定されたわけではない(サイドメモ13:リンチ症候群のスクリーニング検査におけるMSI検査の注意点)。このような場合,その後も既往歴,家族歴に注意を払いながら経過観察を行い,大腸癌に対しては少なくとも3~5年毎に大腸内視鏡検査を行う。
- 「改訂ベセスダガイドラインを満たす」が,「アムステルダム基準Ⅱを満たさない」または「リンチ症候群を強く疑う既往歴・家族歴がない」場合でも,MSI-HまたはMMRタンパク消失であれば,リンチ症候群の可能性がある(多くは散発性大腸癌と考えられる)。既往歴,家族歴に注意を払いながら経過観察を行う。
- 家族歴,既往歴からリンチ症候群の可能性が低いと考えられるMSS/MSI-LまたはMMRタンパク消失なしの大腸癌症例では,特別なサーベイランスは行わず,大腸癌またはその他の関連腫瘍を疑う症状が出現,もしくは血縁者に新たな関連腫瘍が発症した場合は,受診を勧める。
6 遺伝カウンセリングと家族(血縁者)への対応
- 患者本人の他に,家族(血縁者)にも遺伝カウンセリングを行うことが望ましい。
- 第1度近親者(親,子,兄弟姉妹)には疾患について十分な説明を行い,同意を得た上でリスク評価に応じた関連腫瘍のサーベイランスを行う。
- リンチ症候群の関連腫瘍の発症は一般に成年期以降であるので,遺伝学的検査の時期も原則的に成年期以降になる。
1)リンチ症候群であることが確定している患者の家族(血縁者)への対応

図28 リンチ症候群であることが確定している患者の家族(血縁者)への対応
- 病的バリアント保持者であることが確定している,あるいは遺伝学的検査を行っていない血縁者にはリンチ症候群としての関連腫瘍のサーベイランスを行う(図28)。
- 病的バリアントがないことが確認された血縁者については,一般のがん検診を行う(図28)。
リンチ症候群の関連腫瘍のサーベイランス開始年齢に達している血縁者に対しては,サーベイランスの必要性,遺伝子診断の意義についての情報を提供する。遺伝学的検査を受けるかどうかは遺伝カウンセリングを通じて本人の意思で決定する。
2)リンチ症候群が疑われるが,確定診断されていない患者の家族(血縁者)への対応
- 遺伝子診断を実施していない,あるいは実施したがリンチ症候群と確定診断することができなかった患者の血縁者には,家系における関連腫瘍の発生年齢や頻度などを参考に個別のリスク評価を行い,関連腫瘍のサーベイランスを行う。
- リンチ症候群が疑われる患者の血縁者の場合,表19のサーベイランスまたは,その家系で最も若い大腸癌診断年齢より5~15歳若い年齢から,大腸内視鏡検査を行う。









お問い合わせ・事務局
〒102-0075東京都千代田区三番町2 三番町KSビル
| 電話 | 03-3263-8697 |
| FAX | 03-3263-8687 |
| jsccr@secretariat.ne.jp | |
| 業務時間 | 平日9時-18時 |